OpenMMをステップバイステップで 〜Part 8 低分子-タンパク複合体シミュレーション〜
OpenMMの使い方を順番にたどる記事のPart 8です。
Part 7まででタンパク質単体のMDシミュレーションについて、「①準備(前処理)、②実行、③結果の解析」の流れがなんとなくわかりました。第1ステージはクリア!…ということにして、第2ステージ「低分子 - タンパク質複合体」のシミュレーションを試したいと思います。
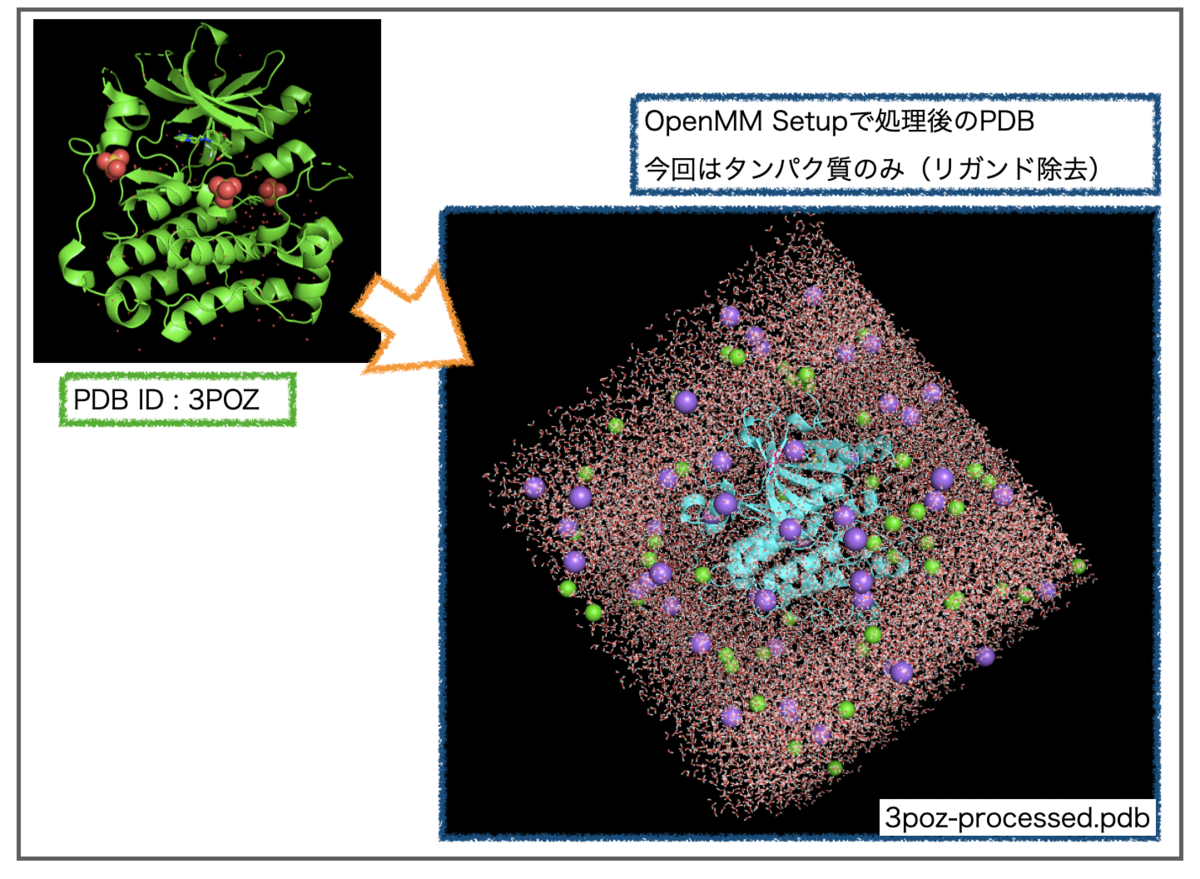
この記事ではTeachOpenCADD T019:Molecular dynamics simulationで行われている「複合体シミュレーション系の構築と計算」の理解を目指します。題材にする構造はPDB ID: 3POZでEGFRタンパクとリガンドの複合体です。
低分子リガンドのための力場としてopen forcefieldを利用し、OpenMMのシミュレーション系を構築します。計算はGoogle Colabで行いました。*1

1. 低分子を含むMDシミュレーションに向けた準備
1-1. タンパク質単独と何が違う?
最初にタンパク質単独と低分子-タンパク質複合体のシミュレーション準備で違う点です。
一番大きな違いは力場です。タンパク質の構造は基本的に(限られた種類の)アミノ酸からなりますが、低分子リガンドはより多様な原子タイプ・結合パターンを含んでいます。なのでより広い領域をカバーした力場パラメータが計算に求められます。
OpenMMにデフォルトで含まれる力場パラメータは生体分子(タンパク質、DNA、RNA ...)や水モデルには対応していますが、低分子には対応していません。そこで別に低分子の力場を作成して取り込んでやる必要があります。
ここではOpenMM Forcefieldsを経由してopen forcefieldの力場を利用します。
1-2. OpenMM Forcefields?
OpenMM Forcefields はOpenMMの追加の力場を提供しているツールです(GitHub)。
サポートしている力場は① AMBER、② CHARMM、③ Open Force Field Initiative force fields です。
低分子についてはAMBERのGeneral AMBER Force Field(GAFF)やOpen Force Fieldのopenff、smirnoffといった力場が使えるようになります。
インストールはcondaでOKです。
conda install -c conda-forge openmmforcefields
1-3. Open Force Field Initiative?
似たような名前が多くて混乱しますが、Open Force Field InitiativeはOpenMMとは別の分子力場に焦点を当てたプロジェクトです。以前こちらのブログ記事でも取り上げました。
継続的な分子力場の改善を目指したイニシアティブで、基になるデータ管理から力場の作成までをオープンかつ再現性よく行うため、自動化した仕組みづくりを目指しているそうです。

現在(2022/05)、以下の3系統の力場が提供されています。

左から右に新しくなります。 SMIRNOFF(スミノフ?)はSMIRKS-native Open Force Fieldの略で、「OpenFF 1.y.x」系統のコードネームがParsley(パセリ)、「OpenFF 2.y.x」系統のコードネームがSage(セージ)です。*2
GitHubへのリンクはそれぞれ以下です。
利用するにはOpen Force Field ToolkitをインストールすればOKです。condaがあれば簡単です。
conda install -c conda-forge openff-toolkit
使える力場パラメータのバージョンは以下で確認できます。
from openff.toolkit.typing.engines.smirnoff import forcefield print(forcefield.get_available_force_fields()) # ['smirnoff99Frosst-1.0.2.offxml', 'smirnoff99Frosst-1.0.0.offxml', 'smirnoff99Frosst-1.1.0.offxml', 'smirnoff99Frosst-1.0.4.offxml', 'smirnoff99Frosst-1.0.8.offxml', 'smirnoff99Frosst-1.0.6.offxml', 'smirnoff99Frosst-1.0.3.offxml', 'smirnoff99Frosst-1.0.1.offxml', 'smirnoff99Frosst-1.0.5.offxml', 'smirnoff99Frosst-1.0.9.offxml', 'smirnoff99Frosst-1.0.7.offxml', 'openff-1.0.1.offxml', 'openff-1.1.1.offxml', 'openff-1.0.0-RC1.offxml', 'openff-1.2.0.offxml', 'openff-1.3.0.offxml', 'openff_unconstrained-2.0.0-rc.1.offxml', 'openff_unconstrained-1.3.1.offxml', 'openff_unconstrained-1.2.1.offxml', 'openff-2.0.0-rc.2.offxml', 'openff_unconstrained-1.0.0-RC2.offxml', 'openff_unconstrained-1.1.0.offxml', 'openff_unconstrained-1.0.0.offxml', 'openff-2.0.0.offxml', 'openff_unconstrained-2.0.0.offxml', 'openff_unconstrained-2.0.0-rc.2.offxml', 'openff-1.1.0.offxml', 'openff-1.0.0.offxml', 'openff-1.0.0-RC2.offxml', 'openff-1.3.1.offxml', 'openff-1.2.1.offxml', 'openff-1.3.1-alpha.1.offxml', 'openff_unconstrained-1.0.0-RC1.offxml', 'openff_unconstrained-1.2.0.offxml', 'openff_unconstrained-1.3.0.offxml', 'openff-2.0.0-rc.1.offxml', 'openff_unconstrained-1.0.1.offxml', 'openff_unconstrained-1.1.1.offxml', 'openff_unconstrained-1.3.1-alpha.1.offxml']
ファイル形式はoffxmlのようです。OpenFFのxmlという感じでしょうか?
OpenFF toolkitがあるとRDKitのMolオブジェクトをOpenFFで使えるように変えたりすることもできます。
1-4. MDTrajのインストール
準備の最後にMDTrajをインストールしておきます。

こちらはTrajとついている通りトラジェクトリ解析用のツールです。前回の記事でご紹介した埼玉大学 松永先生のコロキウム(生体分子系の分子動力学シミュレーションデータの解析入門)でも、ファイルの拡張子変換機能(mdconvert)が使いやすいと紹介されていました。
今回の記事では、別々に前処理したタンパク質と低分子リガンドを一つの複合体にまとめる(マージする)ツールとして利用します。
インストールはcondaでOKです。
conda install -c conda-forge mdtraj
これでツールの準備はおしまいです。
(省略しましたがRDKitも使います。*3)
2. 計算の対象構造について
具体的なシミュレーションの準備を行う前に、計算の対象の構造を確認しておきます。
今回計算の対象とする低分子-タンパク質複合体の構造はTeachOpenCADD T019:Molecular dynamics simulationを踏襲してPDB ID: 3POZです。
EGFRタンパクのキナーゼドメインとリガンド TAK-285のX線共結晶構造です。*4

文献はこちら
Aertgeerts K, Skene R, Yano J, et.al., Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J Biol Chem. 2011(286)18756-65. doi:10.1074/jbc.M110.20619351133-2/fulltext)
複合体構造の中でのTAK-285のLigand IDは03Pです。あとでPDBファイルから構造を取り出す際に使います。*5
3. タンパク質-低分子複合体の系をつくろう!
それでは具体的な処理を進めていきましょう!
この記事では操作の理解を重視するため、TeachOpenCADDでは関数にまとめて定義してある部分を解体して眺めていきます。
3-1. 処理全体の流れ
最初に処理の流れを確認します。以前の記事(part 5 シミュレーションプログラムの中身を理解しよう)で取り上げましたが、OpenMMのシミュレーションプログラムはざっくり以下のようなクラス構成です。
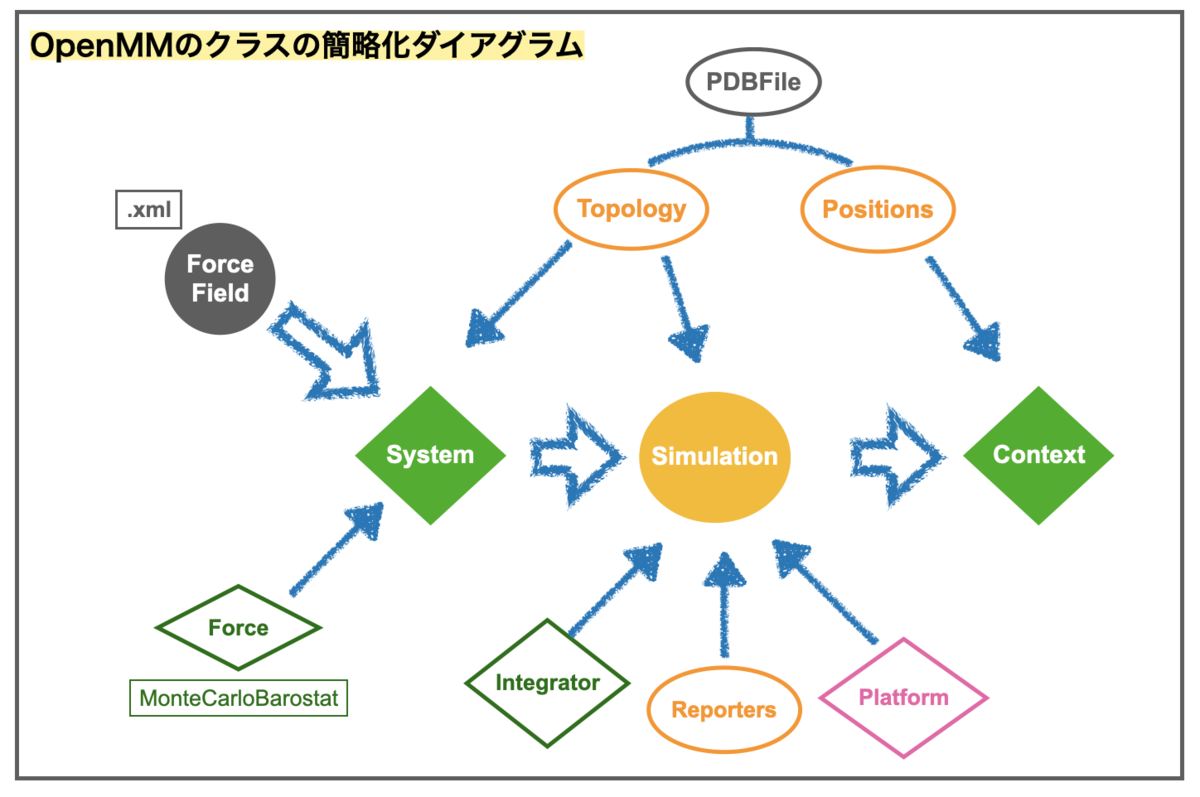
今回、低分子リガンドを加えるにあたって処理を増やすのは左半分です。
追加する処理を加えてみました(水色)。

タンパク質-リガンド複合体のPDBファイルから「①リガンド部分を抜き出し」て、「② Open Forcefieldで力場を設定」し、「③ タンパクとマージ」して複合体に戻します。
より具体的に、処理と使うツールは以下のようになります。
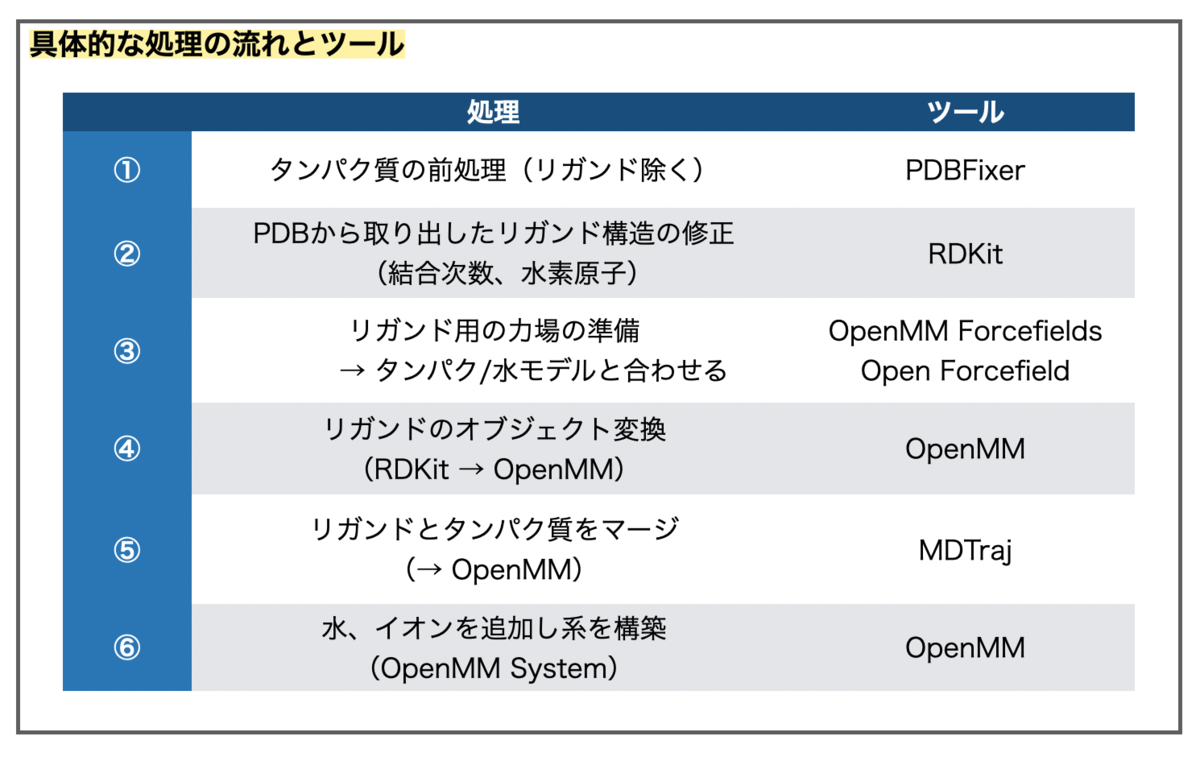
PDBファイルには低分子リガンドの結合次数や水素原子の情報が含まれていないので、これらを補完するためにRDKitを使います。Open Forcefieldを使って低分子の力場を補充し、タンパク-低分子の複合体の系を構築し、最後にシミュレーションの箱(水、イオン追加)を用意して設定完了です。
3-2. Step 1:タンパク質の前処理
Step ①はタンパク質部分の前処理です。ここで行う作業は以前の記事(「Part 2 PDBFixerでタンパク質の前処理」)で取り上げた内容とほぼ同じです。
PDBFixerを使って以下のような作業をします。

水やイオンの追加はここではまだ行わないことにご注意ください*6。
こんな感じのコードです。*7
# 処理したいPDBファイルの指定 pdb_file = 'Data/3poz.pdb' # 必要なライブラリのインポート import pdbfixer # PDBFixerでPDBファイルを読み込む fixer = pdbfixer.PDBFixer(pdb_file) # 不要な構造の削除 fixer.removeHeterogens() # 欠けている残基のチェック(欠損原子の確認のためにも必要) fixer.findMissingResidues() # タンパク質末端の欠けている残基を取り除く処理 chains = list(fixer.topology.chains()) keys = fixer.missingResidues.keys() for key in list(keys): chain = chains[key[0]] if key[1] == 0 or key[1] == len(list(chain.residues())): del fixer.missingResidues[key] # 非標準な残基が含まれているか確認、あれば標準的なものに置き換える fixer.findNonstandardResidues() fixer.replaceNonstandardResidues() # 欠けている原子の確認、あれば追加する fixer.findMissingAtoms() fixer.addMissingAtoms() # 水素原子の付与(pHを設定する) ph = 7.0 fixer.addMissingHydrogens(ph) print(type(fixer)) # <class 'pdbfixer.pdbfixer.PDBFixer'>
得られたPDBFixerのオブジェクトには処理後のタンパク質の原子の座標(positions)とトポロジー(topology)が含まれています。
3-3. Step 2:リガンド構造の修正
続いてリガンド構造の準備です。
PDBから構造や座標を取り出しますが、結合次数や水素原子の情報がPDBファイルには含まれていません。そこでこれらの情報をRDKitを利用して追加します。
まず、RDKitでPDBファイルを読み込んで残基ごとにスプリット(SplitMolByPDBResidues)し、リガンド(ID:03P)を抜き出します。
from rdkit import Chem from rdkit.Chem import AllChem # PDBファイルを読み込んで残基ごとにスプリット rdkit_pdb = Chem.MolFromPDBFile('Data/3poz.pdb') rdkit_pdb_split = Chem.SplitMolByPDBResidues(rdkit_pdb) # 残基 03Pの抜きだし pdb_tak285 = rdkit_pdb_split["03P"] Chem.Draw.MolToImage(pdb_tak285)

単結合だけだと寂しい感じ・・・。
SMILES情報からTAK-285のリファレンス構造を作成し、リファレンスをもとに結合次数を追加しましょう(AssignBondOrdersFromTemplate])。
SMILESはRCSB PDBのリガンドページ(03P)から持ってきました。
# 中途半端に残ってる水素原子を消しておく pdb_tak285_noH = Chem.RemoveHs(pdb_tak285) # SMILESからリファレンス構造を作成 smi_tak285 = "CC(C)(CC(=O)NCCn1ccc2c1c(ncn2)Nc3ccc(c(c3)Cl)Oc4cccc(c4)C(F)(F)F)O" ref_tak285 = Chem.MolFromSmiles(smi_tak285) # リファレンスをもとに結合次数をアサイン prep_tak285 = AllChem.AssignBondOrdersFromTemplate(ref_tak285, pdb_tak285) prep_tak285.AddConformer(pdb_tak285.GetConformer(0)) # 水素原子の付加 prep_tak285 = Chem.AddHs(prep_tak285, addCoords=True) Chem.Draw.MolToImage(prep_tak285)

3-4. Step 3:リガンド用の力場の準備
リガンド構造が用意できたので力場を準備します。
最初にOpenMMでタンパク質(amber14-all.xml)と水モデル(amber14/tip3pfb.xml)の力場を設定しておきます。
from openmm import * from openmm.app import * FF = ForceField('amber14-all.xml', 'amber14/tip3pfb.xml')
このForceFieldに低分子の力場を追加します。
まず、リガンドを「RDKitのMolオブジェクト」から「OpenFFのMoleculeオブジェクト」に変更します。openff toolkitのMolecule.from_rdkitを使えばOKです。*9
from openff.toolkit.topology import Molecule off_tak285 = Molecule.from_rdkit(prep_tak285, allow_undefined_stereo=True, hydrogens_are_explicit=True) print(type(off_tak285)) # <class 'openff.toolkit.topology.molecule.Molecule'>
OpenMMのForceFieldに低分子の力場を追加するには、低分子の構造から① Templateを作ってそれを②ForceFieldにregisterします。
今回はOpen Forcefieldに用意されているGAFFを力場に使います。この場合は「① GAFFTemplateGenerator」と「② registerTemplateGenerator」を使うことになります。
from openmmforcefields.generators import GAFFTemplateGenerator gaff_temp = GAFFTemplateGenerator(off_tak285) FF.registerTemplateGenerator(gaff_temp.generator)
なお、GAFFはAMBER力場の仲間で、低分子医薬品デザインのために作られたものだそうです。タンパク質側をAMBER力場で設定しているときに互換性の良い低分子力場となる感じでしょうか?
今回使用したGAFFのバージョンや、Open forcefieldにインストールされているGAFFのバージョン一覧は以下のように確認できます。
print("version info : ", gaff_temp.gaff_version) print("installed versions : ", GAFFTemplateGenerator.INSTALLED_FORCEFIELDS) # version info : 2.11 # installed versions : ['gaff-1.4', 'gaff-1.8', 'gaff-1.81', 'gaff-2.1', 'gaff-2.11']
使われたバージョン 2.11のようです。
これで力場の準備が終わりました。
3-5. Step 4:リガンドのオブジェクト変換
つづいてリガンドのオブジェクトを変換します。タンパク質とマージするための準備です。
力場を作る際にRDKitからOpenFFのMoleculeオブジェクトにしましたが、これをさらにOpenMMのオブジェクトにします。
まず、OpenFF Moleculeオブジェクトに、分子および原子の名前を追加します。原子の名前は「元素記号 + 数字」形式です。
# 分子の名前("LIG")を追加 off_tak285.name = "LIG" # 原子の名前を追加 # 辞書を用意しておいて、元素記号+数字形式にする element_counter_dict = {} for off_atom, rdkit_atom in zip(off_tak285.atoms, prep_tak285.GetAtoms()): element = rdkit_atom.GetSymbol() if element in element_counter_dict.keys(): element_counter_dict[element] += 1 else: element_counter_dict[element] = 1 off_atom.name = element + str(element_counter_dict[element])
RDKitのGetSymbolで原子の元素記号を一つずつ取り出して辞書に格納しています。同じ元素記号があったら数字を+1するので、炭素なら「C1、C2・・・」と順番に原子に名前がつけられていきます。
名前をつけたらOpenFFからOpenMM(topology、positions)に変換します。ひとつ注意点として、OpenMMの単位はnm基準なので、PDBファイルのオングストロームから変更する必要があります。
# OpenMMの単位系導入 from openmm import unit # トポロジー(topology)の取り出しと変換 off_top = off_tak285.to_topology() omm_top = off_top.to_openmm() # 座標(positions)の取り出し omm_pos = off_tak285.conformers[0] # 単位をÅからnmへ for atom in omm_pos: coords = atom / atom.unit atom = (coords / 10.0) * unit.nanometers # TopologyとPositionsをまとめてOpenMMのオブジェクトに変換 omm_tak285 = app.Modeller(omm_top, omm_pos)
最後にtopologyとpositionsをまとめてOpenMMのModellerオブジェクトにしています。
これでリガンド側での準備はもろもろ完了です。タンパク質と合体しましょう!
3-6. Step 5: リガンドとタンパク質のマージ
ここまででタンパク質のPDBFixerオブジェクトとリガンドのModellerオブジェクトが用意できました。これらをまとめて一つにします。
topologyとpositionsを別々に処理します。
まずtopologyですが、MDTrajを利用します。タンパク質とリガンドをそれぞれMDTrajのオブジェクトに変換(Topology.from_openmm)したのちjoinで結合し、OpenMMオブジェクトに戻します(to_openmm)。
import mdtraj as md # タンパク質とリガンドのトポロジーをそれぞれMdTrajに変換 md_protein_top = md.Topology.from_openmm(fixer.topology) md_ligand_top = md.Topology.from_openmm(omm_tak285.topology) # joinでひとつのトポロジーにまとめる md_complex_top = md_protein_top.join(md_ligand_top) # OpenMMオブジェクトにする omm_complex_top = md_complex_top.to_openmm()
ついで、positionsです。
各原子の位置の値と単位(nm)を含むタプルの配列を作成します。OpenMMのQunatityクラスとなります。
import numpy as np # タンパクとリガンドを合わせた総数 total_atoms = len(fixer.positions) + len(omm_tak285.positions) # positionsを格納するQuantity配列の準備 complex_positions = unit.Quantity(np.zeros([total_atoms, 3]), unit=unit.nanometers) # タンパク質のpositionsを追加 complex_positions[: len(fixer.positions)] = fixer.positions # 後に続けてリガンドのpositionsを追加 complex_positions[len(fixer.positions) :] = omm_tak285.positions
topologyとpositionsがそれぞれ処理できたので、これらをまとめたOpenMMのオブジェクトにしましょう。
comp_model = app.Modeller(omm_complex_top, complex_positions)
できました!
3-7. Step 6:水、イオンを追加し系を構築
前処理の最後として系に水とイオンを追加します。
水のpaddingの厚みは「1.0 nm」、イオンは「NaCl 0.15 M」とします。水モデルはTIP3Pで、forcefiled(amber14/tip3pfb.xml)で指定されています。
comp_model.addSolvent(FF, padding=1.0 * unit.nanometers, ionicStrength=0.15 * unit.molar)
処理後の状態をPDBファイルで保存しておきましょう。
# 処理後の状態(トポロジー、原子の位置)をPDBファイルで出力 top = comp_model.getTopology() pos = comp_model.getPositions() app.PDBFile.writeFile(top, pos, open('Data/gaff_complex_processed.pdb', 'w'))
こんな構造になりました。

4. 計算条件の設定と実行
4-1. 計算条件の設定
計算系の準備が完了したので、シミュレーションを実行するための設定を行いましょう。
タンパク質単独でシミュレーションした過去記事(「Part 3」)と同様の設定にしたいと思います。
こんな感じです。

コードにするとこんな感じです。
from openmm import * from openmm.app import * from openmm.unit import * # シミュレーションの準備 ## ForceFiledからSystemを構築する system = FF.createSystem(comp_model.topology, nonbondedMethod=PME, nonbondedCutoff=1.0*nanometers, constraints = HBonds, rigidWater=True, ewaldErrorTolerance=0.0005) ## 圧力制御の機能をaddForceする system.addForce(MonteCarloBarostat(1.0*atmospheres, 300*kelvin, 25)) ## 積分計算の設定 integrator = LangevinMiddleIntegrator(300*kelvin, 1.0/picosecond, 0.002*picoseconds) integrator.setConstraintTolerance(0.000001) ## 計算環境をPlatformで設定 platform = Platform.getPlatformByName('CUDA') platformProperties = {'Precision': 'single'} ## simulationの構築 simulation = Simulation(comp_model.topology, system, integrator, platform, platformProperties) simulation.context.setPositions(comp_model.positions) # XML結果出力ための設定 with open("system.xml", mode="w") as file: file.write(XmlSerializer.serialize(system)) with open("integrator.xml", mode="w") as file: file.write(XmlSerializer.serialize(integrator)) # エネルギー最小化の実行 print('Performing energy minimization...') simulation.minimizeEnergy() # 平衡化のステップ print('Equilibrating...') simulation.context.setVelocitiesToTemperature(300*kelvin) simulation.step(50000) # シミュレーション本番の実行の前にreporterオブジェクトを追加する print('Simulating...') dcdReporter = DCDReporter('trajectory.dcd', 10000) dataReporter = StateDataReporter('log.txt', 1000, totalSteps=500000, step=True, speed=True, progress=True, potentialEnergy=True, temperature=True, separator='\t') checkpointReporter = CheckpointReporter('checkpoint.chk', 10000) simulation.reporters.append(dcdReporter) simulation.reporters.append(dataReporter) simulation.reporters.append(checkpointReporter) simulation.currentStep = 0 # シミュレーション本番の実行 simulation.step(500000) # シミュレーション最後の状態をXMLで書き出す simulation.saveState("final_state.xml") # 最後の状態をPDBx/mmcifで出力する設定 state = simulation.context.getState(getPositions=True, enforcePeriodicBox=system.usesPeriodicBoundaryConditions()) with open("final_state.pdbx", mode="w") as file: PDBxFile.writeFile(simulation.topology, state.getPositions(), file)
項目が多いので長いですが、前回同様にメインは「①系の構築 → ② エネルギー最小化 → ③ 平衡化のステップ → ④ トラジェクトリのプロダクションステップ」を行っているだけで、間に結果出力(Reporter)の設定が入っています。
それぞれの操作の意味については以前の記事(「Part 5:シミュレーションプログラムの中身を理解しよう」)をご参照いただければと思います。
4-2. TeachOpenCADDの設定との違い
以上の計算条件の設定は基本的にはTeachOpenCADD T19と同じですが以下の点が違います。
- ① アンサンプル:TeachOpenCADDは圧力制御を加えていない
- ② 非結合相互作用の拘束条件(
constraints、rigidWater) - ③ シミュレーション時間 (平衡化の有無、プロダクションの時間)
- ④ 出力ファイル形式の設定
また、TeachOpenCADDでは変数としてforcefieldやmodellerなどを使っています。これらは同じ名前のメソッドがOpenMMに含まれている場合があり、最初のimportで読み込むmoduleを変えると誤認識される恐れがあります。重複のない変数名に変えた方が無難だともいます。
4-3. GoogleColabで実行する際の注意点
以上の計算条件の設定を実行すればMDシミュレーションが行えるはずですが、1nsの計算をするのは大変です。前回(「Part 4:ColabでOpenMMシミュレーションを実行」)と同様に、今回も計算はGoogle Colabで実行しました。
ひとつ注意点として、前回は「①minicondaをインストール」し、そこに「② condaで色々なソフトウェアをインストール」する手順を取りました。
ですが、今回この方法ではRDKitがうまく動きませんでした*10。
回避策としてTeachOpenCADDを参考にcondacolabを利用しました。
こんな感じ。
!pip install condacolab import condacolab condacolab.install() !conda install -q -y -c conda-forge mdtraj openmm openmmforcefields openff-toolkit pdbfixer rdkit
condacolab.install()を実行すると一度「kernel restart」しますが、気にせず次の「!conda install ~~~」に進んでOKです。
より良い方法をご存知の方教えていただけると嬉しいです。
4-4. シミュレーション動画
結果はこんな動画になりました!
めっちゃ動くやん!
解析は次回!
5. おわりに
以上、今回はMDシミュレーションお勉強の第2ステージということで「低分子 - タンパク質複合体」計算系の設定と実行を行ってみました。
…といってもTeachOpenCADD T19の内容をひとつひとつ確かめただけですが、、、余計にわかりにくくなっていたらごめんなさい。
タンパク質単独のシミュレーションと違って、複合体では低分子リガンドの構造情報の取り扱いや専用の力場の設定が必要になりました。そのため複数のツールを組み合わせて行ったり来たりしなければならず難しかったです。
また、低分子力場を扱うためにOpen Forcefieldを使いました。本当はGAFFではなくSMIRNOFFやOpenFFを使ってみたかったのですが、私の能力では無理でした。
以上、今回も色々と間違いが多いと思います。ご指摘いただければ幸いです。
ではでは!
6. おまけ
低分子の力場としてGAFFではなくOpenFFの利用を試みるとこうなりました。
# 力場の設定・追加 from openmmforcefields.generators import SMIRNOFFTemplateGenerator smirnoff_temp = SMIRNOFFTemplateGenerator(off_tak285) forcefield.registerTemplateGenerator(smirnoff_temp.generator) print("forcefield version : ", smirnoff_temp.forcefield) # forcefield version : openff-2.0.0
最新のOpenFF 2.00(Sage)が使われました。
これを使って進めるとOpenMMで系に「水、イオンを追加(addSolvent)」する際に以下のような出力が出ました。
# 〜〜〜略〜〜〜 <Improper class1="[H][O][C]([C]([H])([H])[H])([C]([H])([H])[H])[C]([H])([H])[C](=[O])[N]([H])[C]([H])([H])[C]([H])([H])[N]1[C]([H])=[C]([H])[c]2[n][c]([H])[n][c]([N]([H])[c]3[c]([H])[c]([H])[c]([O][c]4[c]([H])[c]([H])[c]([H])[c]([C]([F])([F])[F])[c]4[H])[c]([Cl])[c]3[H])[c]21$C22#29" class2="[H][O][C]([C]([H])([H])[H])([C]([H])([H])[H])[C]([H])([H])[C](=[O])[N]([H])[C]([H])([H])[C]([H])([H])[N]1[C]([H])=[C]([H])[c]2[n][c]([H])[n][c]([N]([H])[c]3[c]([H])[c]([H])[c]([O][c]4[c]([H])[c]([H])[c]([H])[c]([C]([F])([F])[F])[c]4[H])[c]([Cl])[c]3[H])[c]21$N5#32" class3="[H][O][C]([C]([H])([H])[H])([C]([H])([H])[H])[C]([H])([H])[C](=[O])[N]([H])[C]([H])([H])[C]([H])([H])[N]1[C]([H])=[C]([H])[c]2[n][c]([H])[n][c]([N]([H])[c]3[c]([H])[c]([H])[c]([O][c]4[c]([H])[c]([H])[c]([H])[c]([C]([F])([F])[F])[c]4[H])[c]([Cl])[c]3[H])[c]21$C20#27" class4="[H][O][C]([C]([H])([H])[H])([C]([H])([H])[H])[C]([H])([H])[C](=[O])[N]([H])[C]([H])([H])[C]([H])([H])[N]1[C]([H])=[C]([H])[c]2[n][c]([H])[n][c]([N]([H])[c]3[c]([H])[c]([H])[c]([O][c]4[c]([H])[c]([H])[c]([H])[c]([C]([F])([F])[F])[c]4[H])[c]([Cl])[c]3[H])[c]21$C23#30" periodicity1="2" phase1="3.141592653589793" k1="1.5341333333333336"/> # 〜〜〜略〜〜〜 <Element Improper at 0x13f32bbc0> # 〜〜〜略〜〜〜
エラーではないようですが、improperとなっているのが気になったので今回は採用しませんでした。
推測に過ぎませんが、OpenFFのSMIRNOFFはSMIRKSベースで2面角の力場を設定しているようなので、適切なクエリに該当しない場合improper判定されるのかもしれません。ご存知の方がいらっしゃったらご教示いただければ幸いです。
*1:ロゴは書くソフトウェアのWebサイトより。イラストはいらすとやさんより。
*2:セージで検索すると薬用サルビアが出てきました。ウォッカからハーブ系統に路線変更したんでしょうか?
*3:このブログを読んでくださっている方は既にインストール済みと思います。
*4:3DビューはRCSB PDB提供のNGL Viewer (AS Rose et al. (2018) NGL viewer: web-based molecular graphics for large complexes. Bioinformatics doi:10.1093/bioinformatics/bty419)
*5:PDBで「Ligand ID:03P」をで検索すると他に3RCD、3W2Oがヒットします。見比べても面白かもしれません。
*6:リガンドを入れる前に水を入れると後でぶつかるから?でしょうか??
*7:詳しい中身は過去記事をご参照いただければと思います。
*8:TeachOpenCADDのコードに従って、結合次数を調整した後、再度AddConformerで元の3D座標を追加していますが、これが必要かはよくわかりません。この操作をしなくてもGetConformer().GetPositions()で確認すると座標は変わらなかったです。
*9:TeachOpenCADDでは引数にhydrogens_are_explicit=Trueが設定されていませんが、デフォルトではFalseで自動的にCham.AddHsを実行することになります。あらかじめ水素原子を付加しているのでTrueにしておきました。
*10:インストール後にimport Chemしようとしたら「必要なGLIBCXX(?)が見つからない」といったエラーでimport出来ませんでした
OpenMMをステップバイステップで 〜Part 7 MDAnalysisでトラジェクトリ解析(②)〜
オープンソースのプログラムでMD計算をお勉強する記事のPart 7です。
今回は前回(part 6)に引き続き、MDAnalysisを使ったシミュレーション結果のトラジェクトリ解析(パート②)を行います。また、解析ツールとしてPyTraj も利用します。
使用するデータはEGFRタンパク質(PDB ID: 3POZ)についてOpenMMで1ns計算したものです。
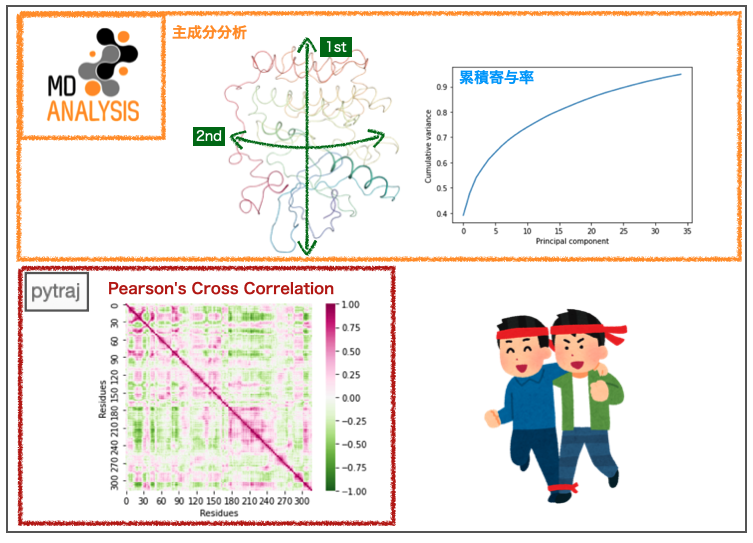
1. この記事で試す解析
前回の記事では①RMSD、②RMSF、③回転半径の計算を行いました。後半戦では④主成分分析と⑤相互相関解析を行います。

ざっくりと「タンパク質のどのあたりが一緒にうごいているか?」、おおまかな動きの特徴をつかめる解析のようです。
2. 主成分分析(PCA)
2-1. MD計算の主成分分析って?
まずは主成分分析です。統計っぽい話でよくみますがMD計算においてはどのように使われるのでしょうか?
前回の記事でも取り上げさせていただいた埼玉大学 松永 康佑先生のコロキウム動画わかりやすく解説してくださっています(動画の33分あたりからです)。*1
こういう感じのお話?
そのままではごちゃごちゃ動いていてよくわからないのが、主成分分析することでノイズを減らして全体としての動きの傾向をつかみやすくなるようです。
上記動画の35分過ぎからは粗視化モデルをつかった主成分分析例が紹介されています。非常にわかりやすいのでおすすめです。
2-2. MDAnalysisで解析の準備(おさらい)
それではMDAnalysisを使った解析を試してみましょう!
前回のおさらいになりますがMDAnalysisはUniverseオブジェクトを中心にデータ解析を行います。topologyとtrajectoryを読み込んで構築します。
import MDAnalysis as mda # topologyとtrajectoryからUniverseオブジェクトを構築 md_universe = mda.Universe("simulation_data/3poz-processed.pdb", "simulation_data/trajectory.dcd") print(type(md_universe))
また、解析に先立ってトラジェクトリのタンパク質のアラインメントをとっておきます(念のため・・)。
from MDAnalysis.analysis import align # トラジェクトリの位置を最初のフレームにリセットする md_universe.trajectory[0] # align.AlignTrajでアラインメントを取る alignment = align.AlignTraj( mobile = md_universe, reference = md_universe, select="protein", in_memory=True) alignment.run()
準備完了!
2-3. MDAnalysisで主成分分析
2-3-1. PCA計算の流れ
主成分分析のやり方については「MDAnalysis User Guide:Principal component analysis of a trajectory」に詳しいチュートリアルがあります。
MDAnalysis.analysis.pca.PCA()を使えば求めることができ、以下のような流れで計算するようです。

それではチュートリアルに従って順番に確認してみましょう。
2-3-2. PCAの実行と累積寄与率の確認
MDAnalysisでPCAを行うにはAnalysisモジュールのMDanalysis.analysis.pca.PCA()を使います。引数にUniverseオブジェクトと目的のAtomGroupの選択基準を渡します。
Cα原子を選択してPCAを行うとこんな感じ。
import MDAnalysis.analysis.pca as pca # Cα原子のPCA Ca_pca = pca.PCA(md_universe, select ='name CA') Ca_pca.run()
PCAの結果の主成分はp_componentsに格納されていて、「原子数 x 3」の配列が成分数分入った配列です(デフォルトで実行した場合は成分数も「原子数 x 3」です。)。
# 主成分を含む配列のサイズ components = Ca_pca.results.p_components print(components.shape) # (951, 951)
シミュレーションの対象としているタンパク質の構造は317残基(Cα原子:317個)なのでちょうど3倍の951になりました。
分散(variance)および累積寄与率(cumulated_variance)を確認してみます。どちらもnumpy.ndarrayなのでインデックスを指定して取り出せます。
# 寄与率 print("1st varinace : ", Ca_pca.results.variance[0]) print("2nd varinace : ", Ca_pca.results.variance[1]) # 累積寄与率 print("1st cumulative varinace : ", Ca_pca.results.cumulated_variance[0]) print("2nd cumulative varinace : ", Ca_pca.results.cumulated_variance[1]) # 1st varinace : 63.373490214896336 # 2nd varinace : 14.112964500184088 # 1st cumulative varinace : 0.39144314860346985 # 2nd cumulative varinace : 0.4786156120633174
第1主成分と第2主成分を合わせた累積寄与率は48%でした。
2-3-3. 累積寄与率のプロット
累積寄与率が95% に達するには何番目まで考慮すれば良いでしょうか?
cumulated_varianceはndarrayなのでnp.where()で95より大きくなる要素のインデックスを取得し、その一番最初を取り出します。
import numpy as np import matplotlib.pyplot as plt # 累積寄与率が95%を超える成分 pc_95 = np.where(Ca_pca.results.cumulated_variance > 0.95)[0][0] print(">95% component number : ", pc_95) # 累積寄与率のプロット plt.plot(Ca_pca.results.cumulated_variance[:pc_95]) plt.xlabel("Principal component") plt.ylabel("Cumulative variance") plt.show() # >95% component number : 35
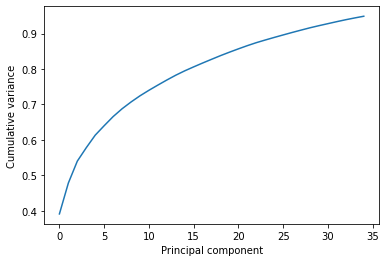
第35主成分まで含めると95%を超えることがわかりました。少しずつ増えていく感じです。
2-3-4. トラジェクトリの次元削減とプロット
PCAを使ってトラジェクトリを次元削減してみましょう。transform()を使うと、指定した数(n_components)の主成分に変換されます。

戻り値はサイズ「(トラジェクトリのフレーム数, 主成分の数)」のndarrayです。
主成分の数を3としてCα原子について変換するとこんな感じです。
# CαについてPCAで次元削減 transformed = Ca_pca.transform(md_universe.select_atoms("name CA"), n_components=3) print("shape of the array : ", transformed.shape) # shape of the array : (50, 3)
2次元にプロットして確認してみましょう。準備としてPandasのデータフレームに変換します。
import pandas as pd # トラジェクトリの1フレームの時間(ps) frame_time = md_universe.trajectory.dt # データフレームに格納する df = pd.DataFrame(transformed, columns=['PC1', 'PC2', 'PC3']) df['Time (ps)'] = df.index * frame_time df.head()
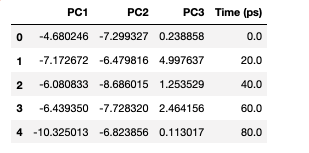
20ps 毎の各フレームで、主成分PC1〜PC3の重みに変換されました。
プロットしてみます。時間推移( Time (ps))で色づけしています。
import matplotlib.pyplot as plt import seaborn as sns pair_grid_plot = sns.PairGrid(df, hue='Time (ps)') pair_grid_plot.map(plt.scatter, marker='.')

PC1とPC2の2次元プロットでは近い色の点が集まった意味ありげなパターンになりました。
・・・何の意味かはわかりません。
2-3-5. 主成分の動きを動画で
PCAを使ったトラジェクトリの基本的な動きの確認には、プロット以外にも動画で確認する方法もあるそうです。
元々のトラジェクトリを各主成分に投影します。
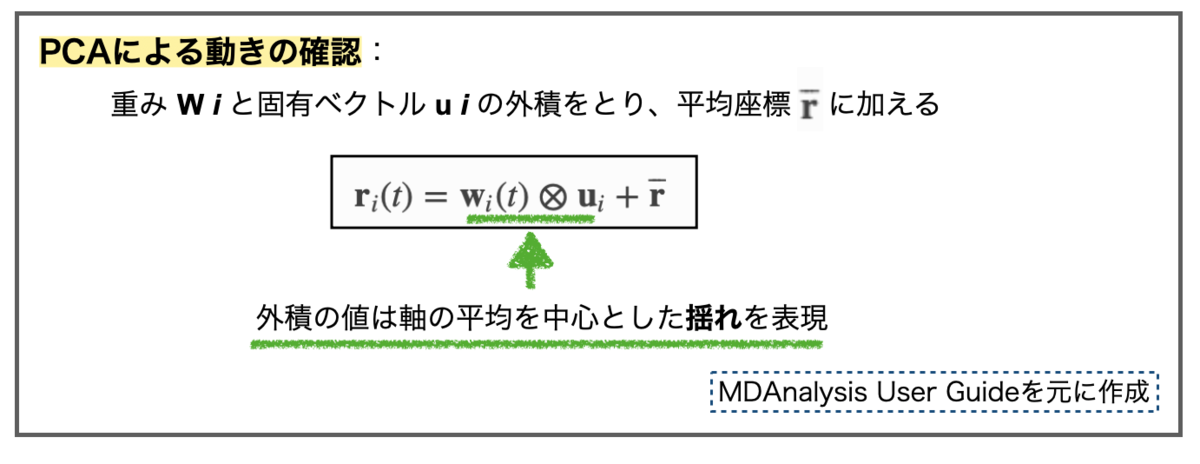
重み(w)は先のtransform()で得られていて、主成分の固有ベクトルはresults.p_components、平均座標はmeanでアクセスできます。上記式を実行しましょう。
# 第1主成分 pc_1 = Ca_pca.results.p_components[:, 0] # 第1主成分についての重み weight_1 = transformed[:, 0] # 平均座標 mean_coord = Ca_pca.mean # 投影 projected_1 = np.outer(weight_1, pc_1) + mean_coord.flatten() # arrayの形を整える coordinates = projected_1.reshape(len(weight_1), -1, 3) print(coordinates.shape) # (50, 317, 3)
最後のreshapeでは、投影を計算後のarrayを「フレーム数、原子数(N)、xyz座標(3)」の次元となるように整えています。
各原子の座標の時間(フレーム)変化のデータが手に入ったので、これをもとにUniverseオブジェクトを作成します。
proj_univ = mda.Merge(md_universe.select_atoms("name CA")) proj_univ.load_new(coordinates, order="fac") # <Universe with 317 atoms>
Merge()で既存のAtomGroupから新しいUniverseオブジェクトを作り、load_newで座標の情報を更新しています。
NGLviewを使ってトラジェクトリの動画を確認しましょう。
import nglview as nv view = nv.show_mdanalysis(proj_univ.atoms) view
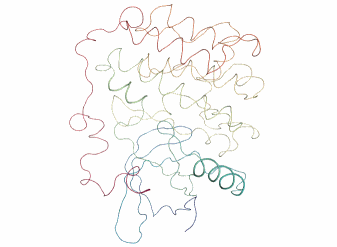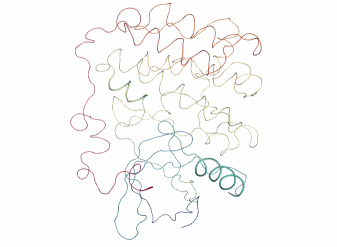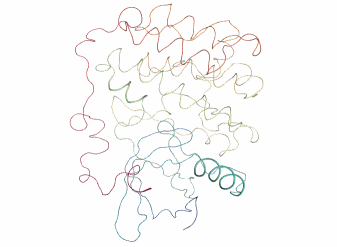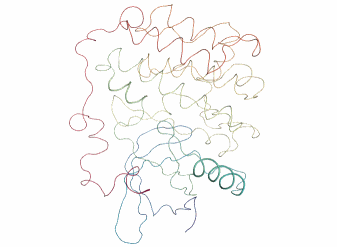
全体が上下方向にクシャッととつぶれるような動きをピクピクしています。
ちなみにPCAで次元削減するまえの元々の動画はこんな感じです。
view = nv.show_mdanalysis(md_universe) view
そのまま見ているよりもPCAの方が動きがシンプルでわかりやすいですね。
PCAのプロットで第1主成分とパターンをつっくていた第2主成分ではどうなるでしょうか?(コードは同じなので省略します。)
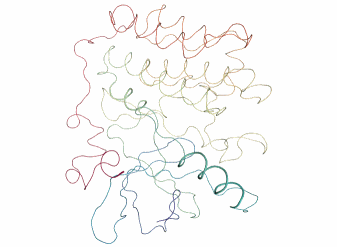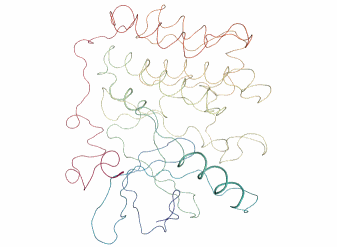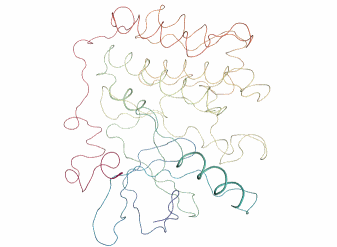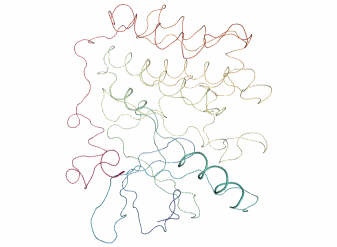
今度は左右方向に揺れている感じです。第1主成分と第2主成分で異なる動きの向きが取り出せていそうです。
楽しい!!・・・意味はわからないですが。
主成分分析は以上です。
3. 相互相関解析(cross correlation)
3-1. 相互相関?
続いて相互相関解析を行います。こういう感じの話です。

こちらも動きの解析ですが、PCAでは基本的な運動のモードを取り出す感じだったのに対し、分子内の運動の相関に焦点を当てているようです。
3-2. PyTrajのインストール
MDAnalysisにそれらしい項目がなかったので、Making it rainで解析に使われていた PyTraj を利用したいと思います。
PyTrajはAMBERのAmber Toolsに組み込まれているcpptrajというプログラムをPythonから扱えるようにしたフロントエンドパッケージだそうです
- GitHub:PyTraj
- ドキュメント:pytraj 2.0.2.dev0
インストールはcondaでできます(チャンネルはambermdです)*2。
conda install -c ambermd pytraj
ただし上記はPython バージョン3.9以上では使えません。Python 3.8以下の仮想環境を作るか、AmberToolsを丸ごとインストールすればPyTrajを使えます*3。
AmberToolsのインストールもcondaでOKです。
conda install -c conda-forge ambertools=21 compilers
こっちはPython 3.9の環境にも入りました。
3-3. PyTrajでトラジェクトリファイルの読み込み
PyTrajで解析を行うためにトラジェクトリを読み込みます。
load()にトラジェクトリファイル(ex. DCD)とトポロジーファイル(ex. PDB)を渡します*4。
import pytraj as pt traj = pt.load("simulation_data/trajectory.dcd", "simulation_data/3poz-processed.pdb") print(traj) # pytraj.Trajectory, 50 frames: # Size: 0.064427 (GB) # <Topology: 57648 atoms, 17895 residues, 17881 mols, PBC with box type = cubic>
なお、PyTrajには他の解析ソフトとの相互利用を意識した機能もあり、MDAnalysisのUniverseオブジェクトからトラジェクトリを読み込むこともできます(PyTraj tutorial:Interface with mdanalysis)。
# MDAnalysisのUniverseオブジェクトから座標をnumpyのarrayに取り出す。 all_atoms = md_universe.select_atoms("all") coords_from_mda = np.asarray([all_atoms.positions for frame in md_universe.trajectory], dtype='f8') # 座標とPDBファイルからPyTrajに読み込む traj_from_mda = pt.Trajectory(xyz=coords_from_mda, top= "simulation_data/3poz-processed.pdb") print(traj_from_mda) # pytraj.Trajectory, 50 frames: # Size: 0.064427 (GB) # <Topology: 57648 atoms, 17895 residues, 17881 mols, non-PBC>
こちらはなぜか周期境界条件が認識されず、non-PBCとなってしまいました。原子数やトラジェクトリのフレーム数など合っていそうです。
3-4. PyTrajでアラインメント
ファイルから読み込んだトラジェクトリでアラインメントをとっておきます。
align()を使います。デフォルトでは全ての原子について最初のフレームを参照構造(ref)にしてアラインメントを取ります。
PyTraj(というよりAmber?)では、原子の選択はmaskと呼ばれるようです(Amber mask selection)。特定の原子団を選択したければ「mask=選択基準」といった形式で引数に渡すことになります。
例えば、Cα原子についてアラインメントを取るには引数mask='@CA'を渡します。
# Cα原子について最初のフレームを参照にアラインメントを取る traj_align = pt.align(traj, mask='@CA', ref=0)
無事、アラインメントが取れました。
3-5. PyTrajで相互相関解析
PyTrajでの解析準備が終わったので相互相関解析を行います。
Matrixのcorrel()を使うことで相互相関の行列が得られます。Cα原子を選択して計算しましょう。
from pytraj import matrix mat_cc = matrix.correl(traj_align, mask ='@CA') print(type(mat_cc)) print(mat_cc.shape) # <class 'numpy.ndarray'> # (317, 317)
計算対象のタンパク質構造の残基数が317なので、「アミノ酸残基 x アミノ酸残基」の2次元行列が得られました。
あとはこれをプロットするだけです。seaborbでヒートマップを可視化します。
ax = sns.heatmap(mat_cc, vmin = -1, vmax = 1, cmap = 'PiYG_r', square=True, xticklabels=30, yticklabels=30) ax.set_xlabel('Residues') ax.set_ylabel('Residues')

① 60 ~ 90(残基番号 760 ~ 790)、② 120 ~ 160(残基番号 820 ~ 860)、③ 180 ~ 240(残基番号 880 ~ 940)あたりに相関が大きくなっている群がありそうです。
PyMolで少し色分けしてみました。

ヘリックスだったりシート構造だったりする部分のようです。「協同運動している箇所」と言われると「そうかも・・・???」という感じですが雑過ぎですね。。。ごめんなさい。
4. おわりに
以上、今回の記事ではMDトラジェクトリの解析(後半戦)を行いました。取り上げた解析は主成分分析と相互相関解析で、MDAnalysisとPyTrajを利用しました。
どちらも揺らぎの大きな特徴をつかむ解析のようですが、主成分分析では運動モードの種類を取り出す感じで、相互相関解析では分子内のアミノ酸残基の協同運動の傾向がでる感じでした。
個人的には第1主成分と第2主成分でそれぞれに投影した動画の運動の向きが変わっていたのが面白かったです。
短いシミュレーションでデフォルトの解析を実行しただけなので生物学的に意味のあることを言えるような記事ではありませんが、とりあえずの使い方がわかったのでヨシ!
以上、今回も間違いが多そうです。ご指摘いただければ幸いです。
ではでは!
OpenMMをステップバイステップで 〜Part 6 MDAnalysisでトラジェクトリ解析(①)〜
OpenMMの使い方を順番にたどる記事のPart 6です。
前回まででOpenMMを使ったMDシミュレーションの準備&実行(とコードの書き方の理解)ができました。今回はシミュレーション結果のトラジェクトリ解析に挑戦したいと思います。
この記事で取り上げるソフトウェアはMDAnalysisです。またコードはTeach OpenCADD T20・Analyzing molecular dynamics simulationsを参考にしています。
途中で力尽きたので今回は解析のパート①です。*1

1. MD計算のデータ解析って何するの?
そもそもMD計算のデータ解析はどんなことをすれば良いのでしょうか?
私のような初学者にとって素晴らしい講演動画と資料がありました!
高度情報科学技術研究機構(RIST)のオンラインサロン 第4回 スパコンコロキウム でおこなわれた埼玉大学 松永 康佑先生のご講演です(研究室HP)。
発表資料(PDF):生体分子系の分子動力学シミュレーションデータの解析入門
①基本的な考え方から、②データ解析入門、③携わられたご研究の実例まで紹介してくださっていてとても楽しい講義です。
特に印象に残った点は「シミュレーション」(データの生成)と「データ解析」の「分業」に分野の研究スタイルが変化しているということです。専用のスパコンで計算したデータが公開され、別のグループがそのデータを使って新規な解析手法の論文化を行う例が増えているそう。オープンサイエンスの力を感じますね。
また、スライドp19「データに溺れないために:データ解析の鉄則」はMDシミュレーションの解析に限らない大事な考え方のように思いました。
紹介されていた研究の実例はこちら。薬剤耐性化に関与する多剤排出トランスポーター AcrBの機構の解明です。

- 論文:Y. Matsunaga, T. Yamane, T. Terada, K. Moritsugu, H. Fujisaki, S. Murakami, M. Ikeguchi, and A. Kidera,
“Energetics and conformational pathways of functional rotation in the multidrug transporter AcrB”,
eLife 7, e31715 (19 pages) (2018) DOI: 10.7554/eLife.31715- 日本語解説:「全原子分子動力学シミュレーションが解き明かす多剤排出トランスポーターAcrBの薬剤排出機構」 生物物理 2019(59)084-087
(小さな)プロトン化状態の変化で(大きな)膜貫通部位の運動がおこるというのは驚きです。タンパク質は本当によくできていますね。
2. この記事で試す解析とパッケージ
タンパク質の動的な挙動の理解はとても魅力的ですが、とても素人に手を出せるものではないので、ここでは基本的な解析手法の動かし方を試すことにします。
具体的には以前このブログでも取り上げたMaking-it-rainを参考に以下の解析を行います。

また利用するMDトラジェクトリ解析パッケージはMDAnalysisです。

MDAnalysisはTeachOpenCADDのT20・Analyzing molecular dynamics simulationsでも使われていて、Pythonから利用できます。また、チュートリアルが充実かつ開発コミュニティが活発そうな感じなのも魅力的です。あとロゴが可愛い。
インストールはpipかcondaでOKです(MDAnalysis - Installation)。
mamba install mdanalysis -c conda-forge
また、トラジェクトリの可視化を行うためにNGLviewを利用します(GitHub, Document)。
こちらもpipかcondaでインストールできます。
mamba install nglview -c conda-forge
mamba速い。
3. シミュレーション全体の様子を眺めよう
3-1. NGLviewerで動画を見よう
冒頭に挙げた松永先生のコロキウムでは、まず最初に「シミュレーションがうまくいったか?動画で眺めること。」を推奨されていました。失敗しているとタンパク質が崩壊するそうです。
動画で確認するために、MDAnalysisを利用してシミュレーション結果のファイルを読み込みましょう。前回の記事(Part 4)で得られたファイルは、「① OpenMMのReporterを使って出力したトラジェクトリのDCDファイル」と「②PDBファイル形式の前処理後の系の構造」です。
MDAnalysisはUniverseと呼ばれるオブジェクトを中心のデータ構造としてさまざまな処理を行う構成になっているようです(MDAnalysis tutorial - Basics)。
Universeオブジェクトはtopologyとtrajectoryを読み込んで構築できます。
import MDAnalysis as mda # topologyとtrajectoryからUniverseオブジェクトを構築 md_universe = mda.Universe("simulation_data/3poz-processed.pdb", "simulation_data/trajectory.dcd") print(type(md_universe)) # <class 'MDAnalysis.core.universe.Universe'>
次に、Jupyter notebook上で動画を確認するためのツールとしてNGL viewerのnglviewを利用します。
nglviewにはさまざまなファイルを可視化する機能があらかじめ用意されており、show_ファイル形式みたいな感じで使えます(nglview : Usage)。
MDAnalysysを使うならshow_mdanalysys()として、Universeオブジェクトを渡せばOKです。
import nglview as nv view = nv.show_mdanalysis(md_universe) view

ぱっと見、大きな崩壊はなさそうですね!よかった。
自動で水やイオンを消して見やすくしてくれるので便利です。
3-2. NGLviewが表示されない?
もしviewを実行してもjupyter notebookに何も表示されない場合は以下をお試しください。
jupyter notebookのバージョンがv5.0の時は原子数の多い大きな系を入力できなくなっている可能性があるので、ノートブック立ち上げ時にフラグを追加して上限を増やしてあげます。
jupyter notebook --NotebookApp.iopub_data_rate_limit=10000000
もしくはwidgetnbextensionやnglviewをノートブックで使えるようにアクティベートする必要があるかもしれません。
NGLviewのGitHub READMEには以下のコマンドが書かれています。*2 *3*4
python -m ipykernel install --sys-prefix jupyter nbextension enable --py --sys-prefix widgetsnbextension jupyter nbextension enable --py --sys-prefix nglview
私の環境では以上を実行すると表示されるようになりました。
3-3. エネルギー変化のプロット
今回のシミュレーションではポテンシャルエネルギー(kJ/mol)や温度(K)の継時的な変化を「log.txt」ファイルに書き出しました。
具体的な解析の前にこちらも確認しておきましょう。タブ区切りなのでPandasのread_table()でOKです。
import pandas as pd log_df = pd.read_table("simulation_data/log.txt") log_df.head()

import matplotlib.pyplot as plt x = log_df["Step"] y = log_df["Potential Energy (kJ/mole)"] plt.plot(x, y) plt.title("change of the potential energy") plt.xlabel("steps (per 0.002ps)") plt.ylabel("potential energy [kJ/mol]") plt.show()

横軸がStepで縦軸が系のポテンシャルエネルギーです。一定の範囲内で揺れ動いているようです。
今回のシミュレーションではStep Sizeを「0.002 ps」として、「500,000 steps」計算しているので1 ナノ秒のシミュレーションです。1000 steps毎にログを出力しているのでデータポイントは2 ps毎の500個になります。
温度が保たれているかプロットしておきましょう。
x = log_df["Step"] y2 = log_df["Temperature (K)"] plt.plot(x, y2) plt.title("change of the temperature") plt.xlabel("steps (per 0.002ps)") plt.ylabel("temperature [K]") plt.show()
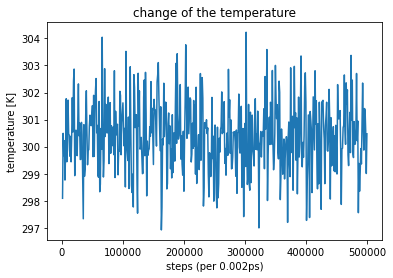
設定の300Kから-4K ~ +4K程度の範囲内に収まっていました。これは十分に温度一定のアンサンブルとみなして良いのでしょうか??
よくわからないので有識者の方ご教示いただけると嬉しいです。
4. MDAnalysisでトラジェクトリ解析
なんとなく上手くいってそう(平衡状態のサンプリングができていそう)なので、MDAnalysisを使ったより具体的な解析に進みます!
4-1. MDAnalysisのオブジェクトと解析対象の選択
まず最初にMDAnaysisにおいて一番基本的なオブジェクトについて触れておきます。
先に述べた通りMDAnalysisでは「topologyとtrajectoryから構築するUniverseオブジェクト」に対してさまざまな操作を行うことで解析を行います。
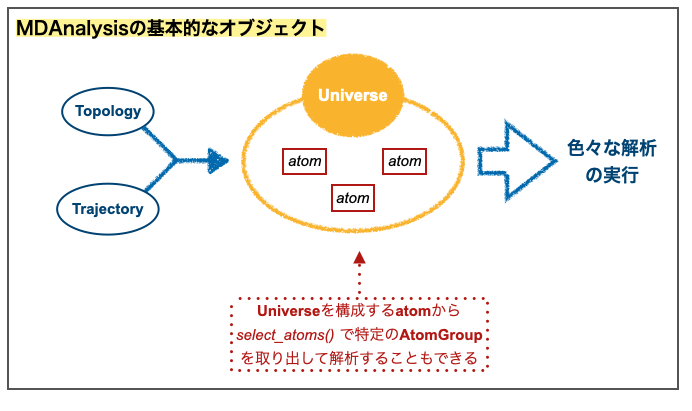
Universeの重要な属性の一つがUniverse.atomsです。これはMDAnalysisの「最も基本的な構成要素であるAtomオブジェクト」のリスト(AtomGroup)です。読んで字のごとく系を構成する原子ですね。
例えば「①Universeオブジェクト、② atomsの最初の一つ、③ atomsの最後の一つ」を取り出してみるとこんな感じです。
print("whole system : ", md_universe) print("example atoms : ", md_universe.atoms[0]) print("example atoms : ", md_universe.atoms[-1]) # whole system : <Universe with 57648 atoms> # example atoms : <Atom 1: N of type N of resname GLN, resid 701 and segid A and altLoc > # example atoms : <Atom 57648: Cl of type Cl of resname CL, resid 20103 and segid 4 and altLoc >
「① 系全部で57648原子」あり、その中には「②タンパク質の701番残基GLNの窒素原子(N)」や「③加えたイオンの塩素原子(CL) 」があることがわかります。
MDシミュレーションのトラジェクトリ解析では「系のうち特定の要素(タンパク質の特定の残基、原子、リガンド)に焦点を当てて時間変化を追跡したい!」といったことが多いようです。
このような時にはMDAnalysis.Universe.select_atoms()メソッドを使うことで、引数で指定した条件のAtomGroupを選択して解析を行うことができます。
例えば「① タンパク質 ("protein")、② 主鎖("backbone")、③ Cα原子 ("name CA")」を選択したい場合は以下のようになります。
print("protein atoms : ", len(md_universe.select_atoms("protein"))) print("backbone atoms : ", len(md_universe.select_atoms("backbone"))) print("CA atoms : ", len(md_universe.select_atoms("name CA"))) # protein atoms : 5120 # backbone atoms : 1268 # CA atoms : 317
それぞれ選択した後lenでatomの数を数えています。「②主鎖の原子数」が「③Cα原子」のちょうど4倍になっているので正しく選択できていそうです。

今回対象としているタンパク質(EGFR)のデータは「PDB ID : 3POZ」です。上図の通り前処理の段階でN末端とC末端を削っているので、シミュレーション構造には317残基残っています。Cα原子の数に一致しているようで良かった!
select_atoms()の引数には他にも色々な選択肢があるようです。ドキュメントの3. Selection commandsなどをご参照ください。
4-2. アラインメント
MDAnalysisの基本的な構成がわかったところで、解析の準備段階として、トラジェクトリにおけるタンパク質のアラインメントをとります。
TeacOpenCADD T20によると、(今回のように)シミュレーション時間が短い時はあまりおこらないそうですが、タンパク質が回転したり移動したりすることがあるそうです。大きく位置がずれると解析が難しくなるのであらかじめアラインメントをとって揃えておく方が良いそうです。
また、MDAnalysisの「ドキュメント: Alignments and RMSfitting」にも、「RMSDやRMSFといった値が意味のあるものにするためにはトラジェクトリのアラインメンとを取ること」との記述があります。
アラインメントにはMDAnalysis.align.AlignTrajを使います(Aligning a trajectory to a reference)。ここではトラジェクトリの最初の構造をリファレンスとして全体を合わせます。
from MDAnalysis.analysis import align # トラジェクトリの位置を最初のフレームにリセットする md_universe.trajectory[0] # align.AlignTrajでアラインメントを取る alignment = align.AlignTraj( mobile = md_universe, reference = md_universe, select="protein", in_memory=True) alignment.run()
動画で見ます。
view = nv.show_mdanalysis(md_universe) view

・・・違いがわからない。
AlingTrajの引数ではmobileで動かす対象のUniverseオブジェクトを指定し、referenceのUniverseオブジェクトに合わせます。上記ではどちらも同じUniverseオブジェクトを指定してますが、実行前にトラジェクトリの最初のフレームにリセットする操作を行なっているため、開始時点の位置に対して全体のアラインメントをとることになっています。
また、引数selectではトラジェクトリのうち「どの構造のアラインメントを取るか」選択することができます。上記では「"protein"」とすることでタンパク質全体を選んでいます。先に見たように「"name CA"」でCA原子(アミノ酸Cα炭素原子)を選択することもできます。
最後に、今回は引数「in_memory=True」とすることでその場でアラインメントをとったものに置き換えていますが、トラジェクトリのサイズが大きくてメモリに載せきれない場合は、一度ファイルに書き出して再度トラジェクトリを読み込むといった処理もできます。
以下のようにすればOKです。
# 検証用にuniverseオブジェクトを作成 md_universe2 = mda.Universe("simulation_data/3poz-processed.pdb", "simulation_data/trajectory.dcd") md_universe2.trajectory[0] # アラインメントをファイルに書き出す場合。 alignment2 = align.AlignTraj(mobile = md_universe2, reference = md_universe2, select='name CA', filename='simulation_data/CA_aligned.dcd', in_memory=False) alignment2.run()
CA原子でアラインメントをとって「CA_alinged.dcd」ファイルに書き出しました。*5読み込んで可視化します。
md_universe3 = mda.Universe("simulation_data/3poz-processed.pdb", "simulation_data/CA_aligned.dcd") view = nv.show_mdanalysis(md_universe3) view

動画のどこが違うがお分かりでしょうか?・・・私にはさっぱりわかりません。
4-3. RMSD
アラインメントも無事とれたので、とっつきやすいRMSD(wikipedia - 原子位置の平均二乗偏差)から解析を行なってみましょう!
だいたいこういう話です。*6

少し注意点ですが、MDAnalysisのメソッドには小文字のrmsdと大文字のRMSDがあり、解析機能(や引数)が異なります。
4-3-1. フレーム2点間のRMSD(rms.rmsd())
まず、RMSDの評価としてトラジェクトリの「フレームの最初と最後でどれだけ構造変化があったか?」を検証してみます。2つの状態の比較の場合はMDAnalysis.analysis.rms.rmsd()を使えば良いようです。rmsdが小文字であることにご注意ください(MDAnalysis Tutorial : 8. Using the MDAnalysis.analysis modules)。
from MDAnalysis.analysis import rms # 最初のフレームにリセット md_universe.trajectory[0] # 最初のタンパク質の位置 protein_first = md_universe.select_atoms("protein").positions # 最後のフレームに移動 md_universe.trajectory[-1] # 最後のタンパク質の位置 protein_last = md_universe.select_atoms("protein").positions # rmsdを評価 print(rms.rmsd(protein_first, protein_last)) # 1.7643614273332058
1.76Å・・・なるほど?・・・この値がどの程度なのか良くわかりません。
4-3-2. トラジェクトリ全体のRMSD推移(rms.RMSD())
次にタンパク質の主鎖(backbone)に焦点を当てて、トラジェクトリ全体での変化を見てみましょう。全体を見る場合はMDAnalysis.analysis.rms.RMSD()を使います(大文字のRMSDに注意!)。
# フレームのリセット md_universe.trajectory[0] # RMSD解析 RMSD_analysis = rms.RMSD(md_universe, # アラインメントを取るオブジェクト md_universe, # アラインメントを合わせるリファレンスのオブジェクト select = "backbone", # 計算対象を主鎖にする ref_frame = 0 # リファレンスのフレームインデックス ) RMSD_analysis.run()
run()で解析を実行すると、結果はresults.rmsdに「[[frame, time(ps), RMSD(A)], [...], [...]]」という情報を含むnumpy.ndarrayの形で保存されています。
print("type : ", type(RMSD_analysis.results.rmsd)) print("1st data : ", RMSD_analysis.results.rmsd[0]) print("total data number : ", len(RMSD_analysis.results.rmsd)) # type : <class 'numpy.ndarray'> # 1st data : [0.0000000e+00 0.0000000e+00 6.0600393e-07] # total data number : 50
matplotlibでプロットしましょう。0 psはリファレンスそのものなので除きます。
time_ps = RMSD_analysis.results.rmsd[1:, 1] backbone_RMSDs = RMSD_analysis.results.rmsd[1:, 2] plt.plot(time_ps, backbone_RMSDs) plt.title("change of the RMSD") plt.xlabel("Time (ps)") plt.ylabel("RMSD [$\AA$]") plt.show()
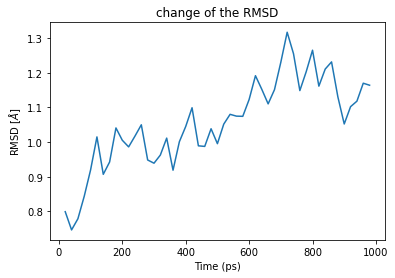
右肩上がりのプロットになりました。600 ps以降は1.1Å前後をうろうろしている感じです。
ついでにRMSDの分布もプロットしてみましょう。seabornのkdeplotを使ってカーネル密度推定法のプロットを行います。
import seaborn as sb sb.kdeplot(RMSD_analysis.results.rmsd[1:, 2], shade=True) plt.title("distribution of the RMSD") plt.xlabel("RMSD [$\AA$]") plt.show()

トラジェクトリ初期のリファレンスとのRMSDが小さい状態は短く、後半のRMSDが1.1Å前後まで広がった状態が多くを占める様子がわかりました。
4-4. RMSF
4-4-1. RMSFって?
つづいてRMSF(平均二乗揺らぎ, Root-mean-square fluctuation)を計算します。これは分子内原子の平均位置からの分散を示す量で、平衡状態からのトラジェクトリ構造の揺らぎを表すそうです。*7

4-4-2. RMSFの計算
RMSFの計算にはMDAnalysis.analysis.rms.RMSF()を使います。引数には計算したいatomgroupを指定します。
アラインメントが取られたタンパク質を使うことが想定された機能なので先のalign.AlignTrajを実行していない場合は処理を忘れずに!
Cα原子についてRMSFを計算する場合はこんな感じです。
# Cαの選択 C_alphas = md_universe.select_atoms("name CA") # RMSF計算 RMSF_analysis = rms.RMSF(C_alphas) RMSF_analysis.run()
run()で解析を実行すると、結果はresults.rmsfにnumpy.ndarrayとして格納されます。RMSDのときと同じですね。
結果をプロットしましょう。Cα原子の残基番号はresnumsで取り出せます。
# 残基番号の取り出し(resnums) residue_numbers = C_alphas.resnums # プロット plt.plot(residue_numbers, RMSF_analysis.results.rmsf) plt.title("C_alpha RMSF") plt.xlabel("residue number") plt.show()
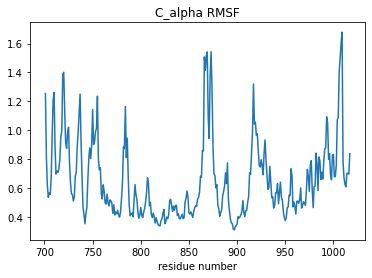
両末端でRMSF(揺らぎ)が大きくなっているのは納得として、850番 ~ 900番にも揺らぎのピークがあります。具体的にはEGFRタンパクのどのあたりの構造なのでしょうか?面白そうなのでもう少し調べます。
4-4-3. 文献計算例との比較
今回のシミュレーションのRMSF解析の結果が妥当か?文献計算例と比較してみましょう。
まず、具体的なタンパク質構造に戻って確認してみます。PyMolで該当の領域(850 ~ 900)を色分けしてみました。
だいたいこの辺。

キナーゼ構造でいうところのActivation loop(A-loop)に相当しそうです。
(ググって最初に出てきた文献と)結果を比較してみましょう。
Tamirat MZ, Koivu M, Elenius K, Johnson MS
Structural characterization of EGFR exon 19 deletion mutation using molecular dynamics simulation.
PLoS ONE 2019(14): e0222814
EGFRの構造を漫画で描くと以下で、DFGモチーフ(D855, F856, G857)に続いてA-loopと呼ばれる構造があるそうです。

上記論文は野生型(Wild EGFR)と変異型(欠損の変異、Δ746ELREA750)についてMDシミュレーションを行なって、変異が構造に与える影響を調べたもののようです。詳細は論文をご参照いただくとして、RMSFの評価を引用します。

野生型、変異型ともにA-loopでRMSFが大きくなっています!また、値としても1〜 2Åということで近い値でした。
素人目には類似の結果が得られているようにみえるのですが、専門家の目からみるとどうなんでしょうか?ご教示いただけると嬉しいです。
4-5. 回転半径(radius of gyration)
最後に回転半径の計算を行ってみます。こういう感じ指標です。*8

回転半径の算出にはAtomGroupに紐づいたメソッドであるradius_of_gyration()を使えば良さそうです。例えばCα原子の回転半径を求めるならこんな感じ。
# フレームのリセット md_universe.trajectory[0] # Cαの選択 C_alphas = md_universe.select_atoms("name CA") # 回転半径の計算 C_alphas.radius_of_gyration() # 19.713873743719788
19.7Åと出ました。select_atoms()で目的の原子群を選択してAtomGroupを作っておいてメソッドを適用するのはこれまで何度か出てきた通りですね。
トラジェクトリ全体を通した変化を見たい場合は、Universeオブジェクトのtrajectory属性についてforループを回してフレームをイテレーションするのが簡単です。*9*10
# フレームを格納するリスト frame_list= [] # 回転半径を格納するリスト rgyr_list = [] # トラジェクトリのフレームごとに回転半径を計算してリストに格納 for ts in md_universe.trajectory: f = ts.frame rgyr = C_alphas.radius_of_gyration() frame_list.append(f) rgyr_list.append(rgyr) # プロット plt.plot(frame_list, rgyr_list) plt.title("change of the radius of gyration") plt.xlabel("frame") plt.ylabel("rgyr [$\AA$]") plt.show()

「1 ns ( = 0.002(ps) x 500,000 (steps))」のシミュレーションが計50フレームなので「1フレーム = 20 (ps) = 10,000 (steps)」です。上下はあるものの途中までは継時的に増加し、19.8Å付近で平衡にあるように見えます。RMSDで見た挙動に似ていますね。
回転半径の分布もプロットしてみましょう。
sb.kdeplot(rgyr_list, shade=True) plt.title("distribution of the radius of gyration") plt.xlabel("rgyr [$\AA$]") plt.show()
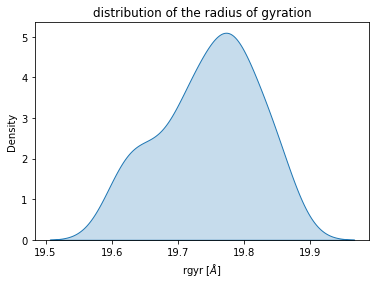
回転半径の分布はRMSDの分布と比較して分布の肩の部分が高くなっています。同じトラジェクトリでも指標によってばらつきの度合いが変わるのは面白いですね。(・・・生物学的な解釈はわかりませんが、、、)
尚、ドキュメント(MDAnalysis User Guide:Radius of gyration)には回転半径を求める関数を自作する例が書かれています。ご興味のある方はご参照ください。
5. おわりに
以上、今回の記事ではMDシミュレーショントラジェクトリの解析を試みました。
冒頭に挙げたコロキウム資料を参考に、①Jupyter notebook上でNGLViewを使った動画の確認を行ったのち、②MDAnalysisを使ったRMSD、③RMSF、④回転半径の算出を行いました。
解析操作で忘れがちな注意点としてはアラインメントをとることとトラジェクトリのフレーム位置のリセットでしょうか?
何はともあれ、素人でもチュートリアルで動かせるOpenMMやMDAnalysisはとても素晴らしいですね!オープンソースコミュニティに感謝です!
個人的にはRMSFでEGFRキナーゼのA-loopの揺らぎが確認できた点が非常に面白かったです。また、RMSDと回転半径で分布が変わってくる点も興味深かったです。
今回取り上げたもの以外にもMDトラジェクトリの解析には色々とあるようです。続きはCM②のあとで!
よく理解せずにまま実行動かしているので間違いが多いと思います。ご指摘いただければ幸いです。
ではでは
*1:下図のMDAnalysisのロゴはドキュメントから。イラストはいらすとやさんより。
*2:NGLviewのDevelopment versionのインストール方法の部分です。
*3: フラッグ--sys-prefixは現在の(アクティベートされてる)仮想環境にkernelをインストールするためのもののようです。
*4:参考:How to properly use --sys-prefix。kernelのインストールがnglviewのアクティベートに必須かはよくわかりませんでした。
*5:引数「in_memory=True」はmobileで指定したトラジェクトリを永久にメモリ上のトラジェクトリ(in-memory trajectory)に変えるもののようです。そのためか、一度align.AlignTrajを実行したUniverseオブジェクトに再度実行してもファイルの書き出しが行われませんでした。そこでここでは再度オブジェクトを作り直しています。
*6:図の記載は理化学研究所 計算科学研究センターのeラーニング「計算生命科学の基礎6 生体系分子シミュレーションの新展開」における横浜市立大学 池口満徳先生(研究室HP)の講義動画(YouTube)とスライド資料(SlideShare)などを参考にしています
*7:こちらも図の記載は先のeラーニング資料などを参考にしています。
*8:こちらも図の記載は先のeラーニング資料などを参考にしています。
*9:参考:MDAnalysis:Overview over MDAnalysis
*10:フレームのイテレーションについてはMDAnalysis UserGuide:Slicing trajectoriesなども参考になります。
OpenMMをステップバイステップで 〜Part 5 シミュレーションプログラムの中身を理解しよう〜
OpenMMの使い方を順番にたどる記事のPart 5です。前回までで、GUIのOpenMM Setupを利用して作成したファイル(①タンパク質構造、②シミュレーション実行プログラム)を用いて、Google Colab上でシミュレーションが実行できることを確認できました。
今回は「シミュレーションプログラムの中身がどのようになっているか?」、 OpenMMのコードの書き方について眺めて見たいと思います。
また、コードの解釈に先立って「OpenMM プログラムの全体像(各クラス間の相関関係)」を把握したいと思います。
- 1. プログラムの全体像(相関図)
- 2. OpenMMのクラスとMD計算上の役割?
- 3. サンプルコードを眺めよう!
- 4. OpenMM Setupで作成したコードを眺めよう!
- 5. クラスの振り返り
- 6. (おまけ)OpenMMのアーキテクチャ
- 7. おわりに
1. プログラムの全体像(相関図)
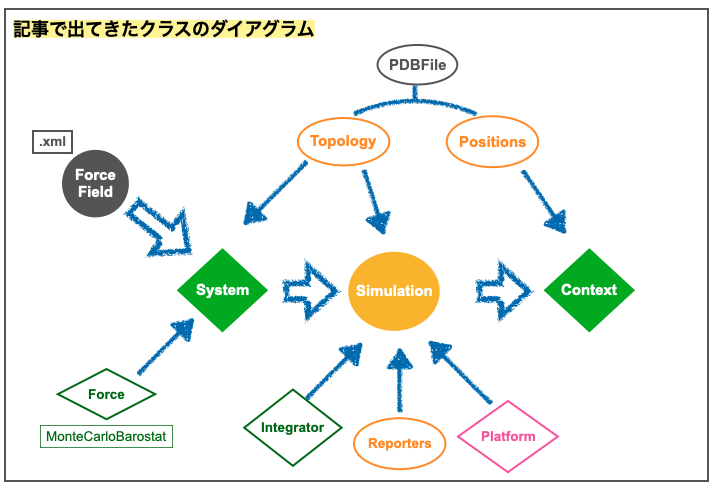
1-1. Diagram of classes in OpenMM 6.0
OpenMMのプログラムの全体像について参考になる図がありました。バージョン 6.0*1についてのものですが、クラス構成は以下のようになっているそうです。

上図はSimTKプロジェクトにあるOpenMMのDocumentsで公開されているOpenMM Class Diagramです。*2
各クラスの図形の意味は左下の点線枠内にあります。
OpenMMはPythonスクリプトで実行できるようになっていますが、内部の仕組みはC++で書かれているものもあります。これらは「API Layer Class」として緑色ひし形でダイアグラムに示されています。SystemやContext、Stateといったクラスです(OpenMM C++ API)。
一方で、Pythonで書かれているものは「App Layer Class」 として黄色円形で示されています。SimulationやReportersなどです(OpenMM Python API)。
また、「App Layer Class」のうちファイルを読み込んで働くものは灰色円形となっています。PDBFileや他のMDソフトで作成した入力(AmberPrmtopFIle etc.)などです。
さらに、各クラスに紐づいたメソッドや生成されるデータは、それぞれ赤枠カードと黄枠カードになっています。
これらのクラスの関係性を矢印でつないでいるのが上図のダイアグラムです。*3
1-2. ちょっと簡略化
ダイアグラムがかなり盛りだくさんなので少し簡略化してみます。

メインになりそうな部分を白抜き表示にしています。それぞれこういう役割のクラスのようです。

説明文だけだとよくわかりません・・・。
2. OpenMMのクラスとMD計算上の役割?
2-1. イメージ
文章だとクラスの役割がわかりにくいので、MDシミュレーションの実行工程とムリやり対応づけてみましょう。
まず、MD計算の流れはこんな感じでした。

これをふまえて、クラスと対応づけるとこんな感じでしょうか???

定義した計算系(System)でシミュレーション(Simulation)を行って、得られた系の時間発展(状態の"文脈", Context)から知りたい時点の情報(State)を適宜取り出すイメージです。
2-2. くどく説明
もう少しくどく書きます。
① まず、シミュレーションを行う系の定義をSystemクラスで行います。「どんなタンパク質?水分子はいくつ?どんな箱に置くの?力場は?…」みたいな基本的な条件設定のイメージです。
② ついで、計算の実行に関する設定項目(「時間発展の積分計算はどうするか?」)や出力方法(「結果のデータやトラジェクトリはどうやって出力する?」)に関する設定を、先の系(System)と結びつけるためにSimulationクラスを利用します。
③ このSimulationを使ってエネルギー最小化(minimizeEnergy())やトラジェクトリの生成(プロダクション、step())を行うことになります。
④ また、MD計算は時間変化を追うシミュレーションなので、系の状態のデータを格納するためにContextクラスが必要になります。例えばシミュレーション開始時点ではタンパク質の各原子の位置情報(pdb.positions)を設定したりします。
⑤ その上で、特定の時点での状態の情報を取り出すためには別途Stateクラスを利用することになります。シミュレーションの要である情報を格納したContextの変数には直接アクセスできない(Stateを経由してアクセスする)仕組みになっているようです。
⑥ 最後に、シミュレーション全体の時間変化やデータはreporterを使って出力することになるようです。
3. サンプルコードを眺めよう!
OpenMMの概観がわかったところで、OpenMM HPにあるサンプルコードを例にPythonの書き方を眺めてみましょう!
3-1. パラメータの復習
コードをみる前に、過去記事でとりあげたOpenMM Setup(GUIツール)で設定したパラメータのイメージを貼っておきます。

たくさんあって大変です。。。
3-2. サンプルコード
サンプルコードではどうなっているでしょうか?
from openmm.app import *
from openmm import *
from openmm.unit import *
from sys import stdout
pdb = PDBFile('input.pdb')
forcefield = ForceField('amber99sb.xml', 'tip3p.xml')
system = forcefield.createSystem(pdb.topology, nonbondedMethod=PME, nonbondedCutoff=1*nanometer, constraints=HBonds)
integrator = LangevinIntegrator(300*kelvin, 1/picosecond, 0.002*picoseconds)
simulation = Simulation(pdb.topology, system, integrator)
simulation.context.setPositions(pdb.positions)
simulation.minimizeEnergy()
simulation.reporters.append(PDBReporter('output.pdb', 1000))
simulation.reporters.append(StateDataReporter(stdout, 1000, step=True, potentialEnergy=True, temperature=True))
simulation.step(10000)
(設定項目の多さから考えれば)10数行でMD計算の実行ができるというシンプルさがOpenMMの良さですね!
3-3. コードの中身
3-3-1. ステップ分割
ではコードの中身を見てみましょう。import部分は省略して、残りは以下のように分割できそうです。
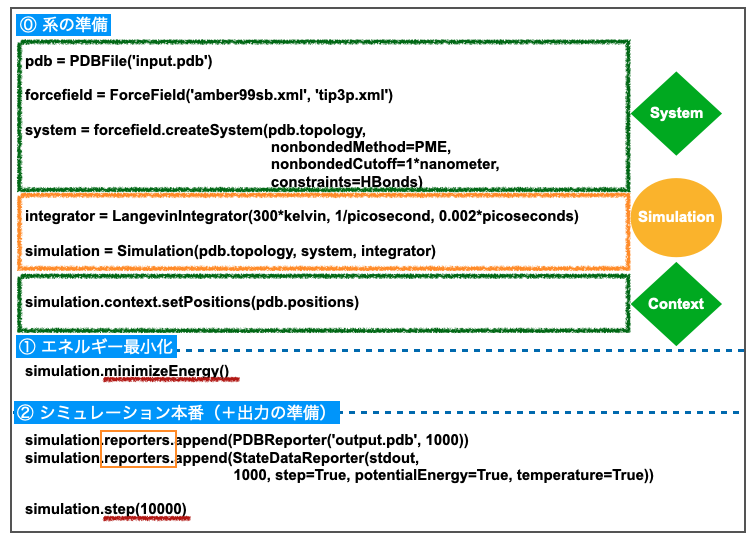
青い点線で「⓪系の準備、①エネルギー最小化、②シミュレーション本番」の3つのステップに区切っています。ここでは「平衡化」に相当する操作がないことにご留意ください。
各コードの右側には「今まで見てきたOpenMMのどのクラスに相当するか?」を書いています。
System、Simulation、そしてContextと順番に構築して「⓪ 系の準備」を行います。ついで「② エネルギー最小化(minimizeEnergy())」を経て「③ シミュレーション本番(step(10000))」を行っています。
データの出力設定(reporters)は「保存したいステップ(③ シミュレーション本番)の直前」に行うというのもポイントになりそうです。
なお、サンプルコードでは状態の確認などを行っていないのでStateクラスが出てこないことにご留意ください。
3-3-2. 系の準備-1
各ステップを細かく見ていきましょう。まずは系の準備です。
最初に、Systemオブジェクトをタンパク質やその他粒子のトポロジーに基づいてForceFieldから構築します。

まず、(水分子の配置などの前処理済みの)PDBファイルの読み込み(PDBFile())と、力場のパラメータを定義したXMLファイルの読み込み(ForceField())を行います。
ここで、ForceFieldは水モデルの力場も合わせて読み込んでいることにご注意ください(amber99sb.xmlとtip3p.xml)*4。
読み込んだらトポロジー(pdb.topology)と、力(相互作用)の計算方法を引数にとってcreateSystem()することでSystemオブジェクトが構築できます。
なお、力の計算の設定は非結合相互作用(粒子・メッシュ・エバルト法、nonbondedMethod=PME)やカットオフ距離、拘束条件などです。
3-3-3. 系の準備-2
次に、Simulationオブジェクトを構築します。これはtopologyとsystem、integratorを引数にとることで構築できます。

Systemの構築に使ったtopologyがもう一度出てくるのが少しわかりづらいです。
積分計算方法(Integrator)としてサンプルではLangevinIntegratorを利用しています。これはランジュバン動力学を利用するもので、温度制御ができます。
LangevinIntegratorの引数は(temperature, frictionCoeff, stepSize)で、サンプルでは熱浴の温度を表す引数に「300*kelvin」が設定されています。
なお、nanometerやkelvinといった単位が利用できるのは最初に以下を実行しているからです。
from openmm.unit import *
単位系が使えるのは便利なので忘れずに実行したいですね。
3-3-4. 系の準備-3
系の準備の最後はsimulationからのcontextの構築です。
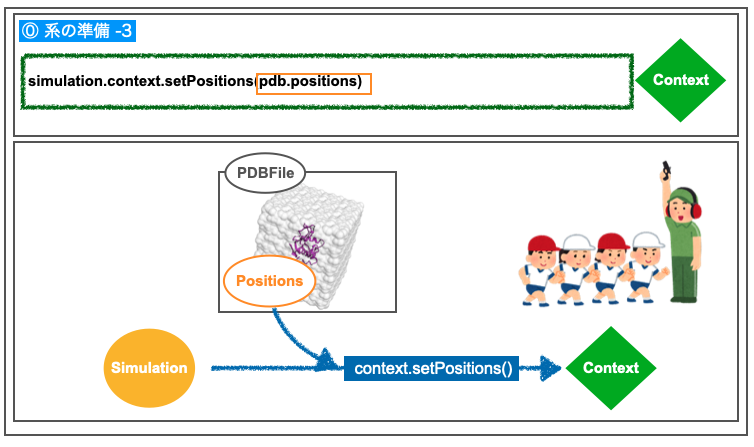
ここではPDBファイルの情報のうち「位置 pdb.position」を利用します。
以上で系の準備は終わりです。
3-3-5. エネルギー最小化
ついで、系のエネルギーを最小化します。

minimzeEnergy()をsimluationに行うだけです。シンプル!
3-3-6. シミュレーション本番
最後にシミュレーション本番です。トラジェクトリのプロダクションです。

step()により、指定したタイムステップのシミュレーションが実行できます。
ただし、ここではトラジェクトリを出力したいので、時間発展させる前に種々のreporterオブジェクトをreporters.append()によりsimulationに追加しています。
出力内容としてPDBReporter()は指定したファイルパスに、一連のフレームを(一定間隔ごとに)PDBファイルとして書き出します。
また、StateDataReporter()はエネルギーや温度といった状態の情報を書き出します。
それぞれの細かな設定はマニュアルをご参照ください。
以上がサンプルコードの中身でした。
4. OpenMM Setupで作成したコードを眺めよう!
4-1. サンプルコードとの違いの概略
サンプルコードを見ることで、シミュレーション実行のためのコードの流れがつかめました。
もう少し長いコード例として、過去記事でOpenMM Setupで作成したものを眺めてみましょう!
コード全体の流れは、予めオブジェクトの作成やメソッドのための色々な引数を変数に代入する形で定義しておき、後ほどシミュレーション実行コードを書くという順番です。
また、サンプルコードのシミュレーションとの大きな違いとして以下の点が挙げられます。
- ① NPTアンサンブルで圧力制御も行う
- ② シミュレーション本番前に平衡化のステップも行う
細かな違いとしては以下の点が挙げられます。
- ③ Reporterの種類の違い
- ④ 計算機環境(platform)の設定を明示的に行っている
4-2. コード全体を眺めよう
違いをふまえた上でコード全体を眺めましょう!
コードの途中にコメントアウトする形で説明を追記しました。
# 必要なライブラリのインポート from openmm import * from openmm.app import * from openmm.unit import * # ファイルの読み込み(PDBと力場XML → Systemのベースになるファイル) pdb = PDBFile('3poz-processed.pdb') forcefield = ForceField('amber14-all.xml', 'amber14/tip3pfb.xml') # Systemの構築に使うパラメータ変数の設定 ## 非結合相互作用の計算方法や拘束条件の定義 nonbondedMethod = PME nonbondedCutoff = 1.0*nanometers ewaldErrorTolerance = 0.0005 constraints = HBonds rigidWater = True constraintTolerance = 0.000001 # 積分計算(Integrator)の設定に必要なパラメータ変数の設定 ## ここではLangevinMiddleIntegrator dt = 0.002*picoseconds temperature = 300*kelvin friction = 1.0/picosecond # アンサンブルNPTとしているため圧力制御も行う(サンプルコードとの違い) ## 圧力制御のパラメータ変数を設定 pressure = 1.0*atmospheres barostatInterval = 25 # シミュレーションを行うための設定 ## シミュレーション時間の設定 ## 平衡化も行うため別途タイムステップを定義している(サンプルコードとの違い) steps = 500000 equilibrationSteps = 50000 ## 計算環境をPlatformで設定している(サンプルコードとの違い) platform = Platform.getPlatformByName('CUDA') platformProperties = {'Precision': 'single'} ## 予めreporterとして何を使うかも変数に定義している dcdReporter = DCDReporter('trajectory.dcd', 10000) dataReporter = StateDataReporter('log.txt', 1000, totalSteps=steps, step=True, speed=True, progress=True, potentialEnergy=True, temperature=True, separator='\t') checkpointReporter = CheckpointReporter('checkpoint.chk', 10000) # シミュレーションの準備 ## ForceFiledからSystemを構築する print('Building system...') topology = pdb.topology positions = pdb.positions system = forcefield.createSystem(topology, nonbondedMethod=nonbondedMethod, nonbondedCutoff=nonbondedCutoff, constraints=constraints, rigidWater=rigidWater, ewaldErrorTolerance=ewaldErrorTolerance) ## 圧力制御の機能をaddForceする(サンプルコードとの違い) system.addForce(MonteCarloBarostat(pressure, temperature, barostatInterval)) ## 積分計算の設定 integrator = LangevinMiddleIntegrator(temperature, friction, dt) integrator.setConstraintTolerance(constraintTolerance) ## simulationの構築 simulation = Simulation(topology, system, integrator, platform, platformProperties) simulation.context.setPositions(positions) # XML結果出力ための設定 with open("system.xml", mode="w") as file: file.write(XmlSerializer.serialize(system)) with open("integrator.xml", mode="w") as file: file.write(XmlSerializer.serialize(integrator)) # エネルギー最小化の実行 print('Performing energy minimization...') simulation.minimizeEnergy() # 平衡化のステップ(サンプルコードとの違い) print('Equilibrating...') simulation.context.setVelocitiesToTemperature(temperature) simulation.step(equilibrationSteps) # シミュレーション本番の実行の前にreporterオブジェクトを追加する print('Simulating...') simulation.reporters.append(dcdReporter) simulation.reporters.append(dataReporter) simulation.reporters.append(checkpointReporter) simulation.currentStep = 0 # シミュレーション本番の実行 simulation.step(steps) # シミュレーション最後の状態をXMLで書き出す simulation.saveState("final_state.xml")
ずいぶん長くなっているように見えますが、実際の操作としてはサンプルコードとそれほど変わっていなさそうです。
4-3. コードの違いを少し詳しく
折角なのでサンプルコードと違う部分を少し詳しくみてみましょう。
4-3-1. NPTアンサンブル(圧力制御)
まず、アンサンブルの違いです。
NPTアンサンブル達成のためMonteCarloBarostatという圧力制御を系に加えています。これは、Systemを構築後にaddForce()の形で追加することで利用可能です。

温度制御に関わるIntegratorがSimulationを構築する際の引数であったのと異なり、その前の「系の構築段階で力として加える」という点は要注意です。
4-3-2. プラットフォーム
続いて計算機環境の設定です。Platformクラスを利用して設定しています。
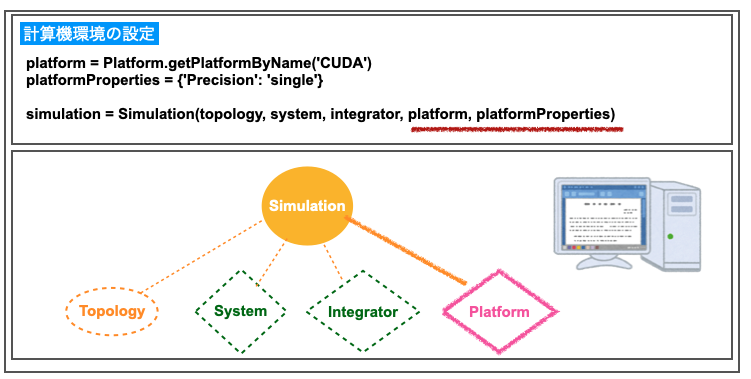
ここではGoogle ColabのGPU(CUDA)を利用するためgetPlatformByName('CUDA')となっています。
Platformはそのプロパティ({'Precision': 'single'})と合わせてSimulationを構築する時に引数として与えられています。
Simulationが計算の実行や出力のさまざまなオブジェクトを取りまとめる役割だったことを考えると納得できそうです。
4-3-3. 平衡化
最後に平衡化ステップを加えることによる違いです。
平衡化の計算自体はシミュレーション本番と同様に、simluationに対して必要な時間に相当するstep()を行えばOKです。
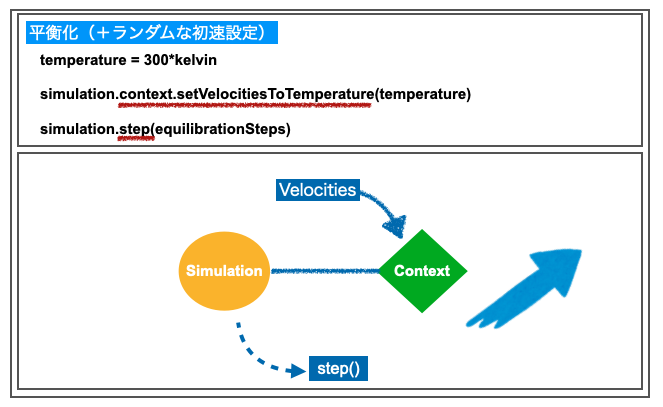
ただし上図のように、OpenMM Setupで作成したコードではさらにsetVelocitiesToTemerature()というものをcontextに与えています。
これは「系の全粒子に対して、設定の温度のボルツマン分布に従うようにランダムな速度を与える」という役割を担います。エネルギー最小化後の系に初速を与えたと考えればよさそうです*5。
contextが速度を含む系の状態を保持するクラスだったことをふまえれば、このような追加方法になるのも納得できそうです。
また、平衡化のstep()の計算が終わったのち、シミュレーション本番のstep()に入る前に以下のコードを実行しています。
simulation.currentStep = 0
平衡化の状態は保持しつつ「ステップのインデックス(currentStep)をゼロ」にすることで、シミュレーション本番のトラジェクトリ出力(reporter)に合わせていると考えられそうです。
以上がコードにおける主な違いでした。
5. クラスの振り返り
サンプルコード、およびOpenMM Setupで作成したコードの振り返りが終わりました。
記事全体のまとめも兼ねてコードにでてきたOpenMMクラスのダイアグラムを作ってみましょう。
こんな感じでしょうか?
一番最初に引用したOpenMM Class Diagramの半分くらいは抑えられたかな?、という印象です。
設定項目とクラスの種類が多いので関係性がわかるとコードを書きやすくなりそうですね。
6. (おまけ)OpenMMのアーキテクチャ
最後に脱線ですが、OpenMMの構成(アーキテクチャ)の図を引用しておきます*6。

OpenMMは計算を実行するためのコードの部分では、計算機の環境(プラットフォーム)を意識しなくて良いように設計されているそうです。
利用者はアーキテクチャ上部のPublic Interface(Public API)の使い方を把握しておけばよく、これはプラットフォームと独立なコードになっています。このPublic APIが上で見たSystemやContextなどです*7。
プラットフォームに依存した部分は、アーキテクチャー下部のPlatform Abstraction Layer(Low Level API)やComputational Kernelsです。最下層が実際に計算を実行するハードウェアに関する部分だそうです。
独自の計算手法を開発する場合などは下層の理解が必要となるそうですが、そもそもそんな上級者はこんなブログ読みませんよね。。。
以上のように、プラットフォーム依存の部分と独立の部分に分かれた構成となっているので、(貧弱な)自分のパソコン上でもGoogle ColabのパワフルなGPUを使った計算のためのプログラム作成が簡単にできたのでしょうか???
よくわかりませんがアーキテクチャって言ってみたかっただけです。ごめんなさい。
7. おわりに
以上、今回はOpenMMでシミュレーションを実行するためのコードの書き方について、「① (OpenMM HPの)サンプルコード」と「② OpenMM Setupで作成したコード」を例に眺めてみました。
また、コードの中身を眺めるに先立って、OpenMM Class Diagramを利用して各クラス間の相関関係を大まかに把握し、OpenMMプログラム全体の概観の理解を試みました。
OpenMMは(ばりばりの計算の専門家でなくても)シミュレーションを実行できるように、Pythonで簡単にシミュレーションプログラムをかけるよう設計されているそうです。
実際に、MD計算に必要な設定項目の多さと多段階工程のややこしさからすれば、シンプルなコードを並べるだけで計算を実行できるようになっていました。
・・・とはいうものの、やはりある程度は複雑になってしまいます。
今回の記事で見たように、クラスダイアグラムのような形式で各操作の関係性がわかっていると動作するコードをちゃんとかけるようになるかもしれませんね?
以上、今回も間違いが多そうです。ご指摘いただければ幸いです。
ではでは!
*1: Class Diagram資料の公開日付は2014年のようです。2022年現在はOpenMM 7.7が最新版となっています。
*2:SimTKは医学生物学計算コミュニティのための無料のプロジェクトホスティングプラットホームだそうです(Whit is SimTK?)。
*3:Lee-Ping Wang博士によるワークショップ(YouTube)「Creating and Customizing Force Fields in OpenMM」での説明などを参考にしています。
*4:利用可能な力場のXMLファイルについてはOpenMM User Guide : 3.6.2. Force Fieldsなどを参照してください。
*5:サンプルコードで初速を設定していない理由はよくわからないです…。NPTアンサンブルのMonteCarloBarostatにも温度がある関係?
OpenMMをステップバイステップで 〜 Part 4-2 よくある質問(FAQ@GitHub)〜
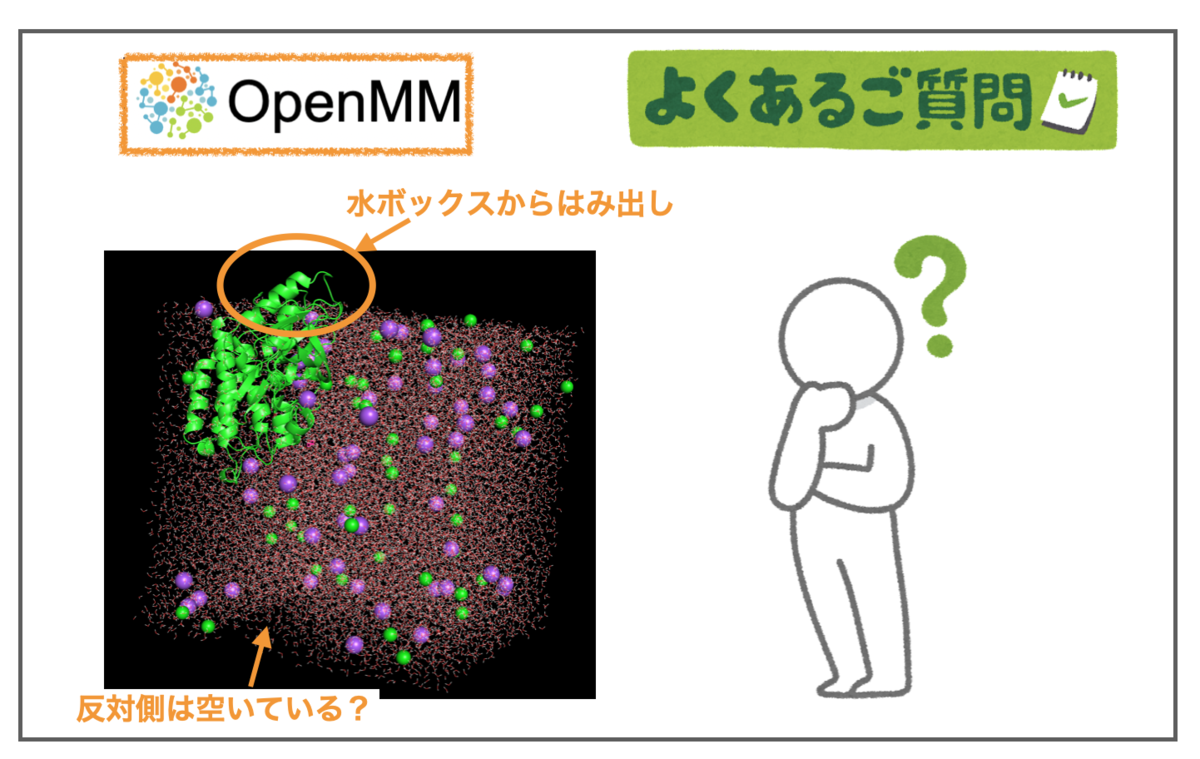
OpenMMのシミュレーションを実行した前回の記事(part 4)で、結果の描画でタンパク質が境界から飛び出ているのが気持ち悪いと書きました。
OpennMMのGitHubページ「Wiki: Frequently Asked Questions」に全く同じ内容が書かれていました。全く気づいていませんでした。。。
よくある質問なら共有しておいた方が良いよね!ということで、DeepL翻訳(にして修正)したものを置いておきます。*1*2
- 1. 周期境界条件はどのように機能するの?
- 2.「粒子の位置がNaNです」「エネルギーがNaNです」といったエラーが出るのはなぜ?
- 3. 「残基のテンプレートが見つかりません」というエラーが出るのはなぜ?
- 4. SystemやForceに加えた変更が無視されるのはなぜ?
1. 周期境界条件はどのように機能するの?
非常によくある質問は、もとを探ると周期的境界条件の理解に関するものです。「なぜ水分子が周期境界条件の箱の外側に拡散するのか?」「なぜタンパク質が水のボックスの端からはみ出るのか?」といった質問がよくあります。 また、「特定の力が周期境界条件においてどのように適用されるか?(あるいは適用されないか?)」といったことに関する質問もあります。 一見、簡単な質問に見えますが、見た目よりもずっと複雑な問題なので、まず初めに説明しましょう。
概念的に言えば、周期境界条件でシミュレーションを行う場合、すべての「粒子」は、空間全体で繰り返しパターンをとる無限の粒子のグリッドを表します。 周期的なボックスが10nmの立方体で、ある粒子が(1, 2, 3)にあるとすると、(11, 2, 3)、(21, 2, 3)、(-9, 12, 103)・・・にも粒子があることになります。これらの位置のうち、どの位置をリストアップするかは重要ではありません。 どれをとっても、全く同じ粒子の無限のグリッドを表しています。
次に、理解すべき非常に重要なポイントがあります。
周期境界条件は、位置ではなく、変位量(displacement)に適用されます。
もう一度10nmの立方体を例にとりましょう。(1, 0, 0) と (9, 0, 0) の2つの粒子間の変位量はどうなるでしょうか? 周期境界条件がない場合、変位量は(8, 0, 0)となります、周期境界条件がある場合、(-2, 0, 0)となります。 もし、2番目の粒子の位置が(-1, 0, 0)や(99, 0, 0)であったとしても、それは問題ではありません。変位量は同じになります。 周期境界条件は、粒子間の変位量に依存する力を計算するときに適用されます。
もう一つ、非常に重要なポイントがあります。
周期境界条件は、非結合相互作用のみに適用され、結合相互作用には適用されません。
2つの粒子間の結合を考えてみましょう。それぞれの粒子は実際には無限の粒子のグリッドを表していますが、だからといって、最初の粒子のすべてのコピーが2番目の粒子のすべてのコピーに結合しているわけではありません。 最初の粒子の各コピーは、2番目の粒子の1つの特定のコピーと結合しており、それ以外のものは結合していません。 もし第2の粒子の異なるコピーが、結合しているコピーよりも何らかの方法で近づくことができたとしても、結合が突然「切れて」異なるコピーと結合をつくりなおすはずだということにはならないのです。
(実際には、本当に必要であれば、結合相互作用に周期境界条件を適用することは可能です。結合相互作用に対して setUsesPeriodicBoundaryConditions() を呼び出すだけでできます。 ですが、通常は行わないことで、特別な状況でのみ適切です。 例えば、一方の端がもう一方の端の次の周期的なコピーに結合しているような無限分子をシミュレートしたいといった場合があげられます。)
これは拘束条件や、実際には結合相互作用の一種となっている非結合相互作用の例外にも適用されます。非結合相互作用は通常、互いに結合している粒子間(1-2、1-3、1-4相互作用)では除外されるか、縮小されます。 これは直接結合している粒子にのみ適用され、その粒子の他の周期的なコピーには適用されません。
その結果、2つの異なる周期的なコピーの間で分子を壊してはいけないということになります。先に、粒子が(9, 0, 0)にあるか(-1, 0, 0)にあるかは重要でないと書きました。しかし、もしその粒子が(1, 0, 0)にある粒子と結合しているならば、それは重要なことなのです。 一方は結合長が8であることを示し、もう一方は結合長が2であることを示します。ほとんどのMDパッケージはこれと同じ要件を満たしているので、通常は問題ありません。 しかし、時々、すべての原子が独立して1つの周期ボックスにまとめられているファイルに出くわすことがあり、分子が真っ二つになっていることがあります。 MDTrajのmake_molecules_whole()関数など、このようなファイルを修正するためのツールがあります。
ここまではシミュレーションの実行に関することでしたが、次に出力ファイルの保存について説明します。 MDエンジンにとって、(0, 0, 0)を中心とする分子は(100, -30, -80)を中心とする分子と全く同一かもしれませんが、トラジェクトリを可視化しようとする場合、異なる分子が多くの異なる周期的な箱に散らばっていると何が起こっているか分からなくなってしまいます。 このため、Context.getState() には、返されたすべての粒子の位置を一つの周期的なボックスにまとめるオプションがあります。 同様に、レポーター関数(reporter)は周期系のトラジェクトリを書き出すときに自動的にこれを行います。ボックスにまとめる作業は常に分子全体に対して行われます。すべての分子の中心は1つのボックスの中にありますが、その分子のいくつかの原子はその外側にはみ出していることがあります。これが、タンパク質の一部だけが水に囲まれているように見えることがよくある理由です。
次のような重要なポイントがあるため、可視化はさらに複雑になっています。
一意に定義された「周期的なボックス 」は存在しない。
例えば、ボックスの幅が10である場合、粒子の位置はすべて0と10の間にあるべきということでしょうか? それとも-5から5の間でしょうか?どちらの方法も様々なプログラムで共通する慣習になっています。 タンパク質が原点を中心とする初期構造を読み込んで、すべての分子を0から10の間に置くようなトラジェクトリを書きだすと、タンパク質のほとんどが水の外に出てしまい、反対側の水のボックスに大きな穴が開いているように見えます。 これは非常によくある問題なので、多くの解析プログラムには特にこの問題に対処する機能が備わっています。例えば、MDTraj の image_molecules() 関数や Cpptraj の autoimage コマンドなどがあります。これらのツールは、すべての粒子を一つの箱に入れて、溶質を中心に、その周りに水を配置するような賢い方法をとります。
2.「粒子の位置がNaNです」「エネルギーがNaNです」といったエラーが出るのはなぜ?
NaNとは「not a number」の略で、「数字ではない」という意味です。NaNは、ゼロで割ったり、負の数の平方根を取ったりするような、実数値がうまく定義できない計算の結果をコンピュータが表現する方法です。このエラーが発生した場合、一般的にはシミュレーションが破綻(blown up)していることを意味します。このようなエラーは様々な理由で発生しますが、一般的には力や速度が大きすぎることが原因です。その結果、粒子が急激にジャンプし、次のタイムステップでさらに大きな力が発生し、数ステップですべての粒子の位置がNaNに変わってしまいます。
シミュレーションが破綻する原因には、さまざまな種類の問題があります。より一般的なものをいくつか紹介します。
タイムステップが大きすぎる場合。 タイムステップが大きすぎる場合、シミュレーションはしばらくの間、正常に実行されたように見えますが、その後、破綻します。タイムステップが大きすぎる場合、すぐに不安定になります。
初期コンフォメーションがエネルギー最小化されていない場合。 例えば、結晶構造を初期コンフォメーションとして使用している場合、座標が力場の最小値になっていない可能性があり、最初のタイムステップで非常に大きな力を発生させます。これは通常、局所的なエネルギー最小化を実行することで修正することができます。
周期的なボックスのベクトルが正しく設定されていない場合。 例えば、Amberのprmtopとinpcrdファイルから開始する場合によくある間違いです。どちらのファイルも周期的ボックスのベクトルを指定しており、お互いに異なる場合があります。粒子座標があるボックスサイズを想定していても、シミュレーションが異なるボックスサイズを使用している場合、粒子間の激しい衝突を引き起こす可能性があります。
拘束条件が正しく定義されていない場合。 拘束条件の適用には行列を逆行列にする作業が含まれます(明示的な場合もあれば、暗黙的な場合もあります)。この行列の条件が悪いと、シミュレーションが破綻する原因になります。単純な原因としては、同じ粒子ペアに2つの異なる拘束条件が課されている場合が挙げられます。また、同時にすべてを満たすことができない矛盾した拘束条件がある場合にも起こりえます。その他、より複雑な拘束条件のセットも、条件付けが悪い行列につながる可能性があります。
力の定義が正しくない場合。 これは、系(System)を直接構築しているとき、あるいは系に余分な力を追加しているときに、最も起こりやすい問題です。適用する力が大きすぎる場合、シミュレーションが破綻する可能性があります。また、カスタマイズした力を作成する際には、直接NaNを生成するような式を書かないように注意してください(負の値を取りうるの値の平方根を取る式など)。
このようなエラーは様々な原因で発生するため、デバッグが困難な場合があります。 最初に確認することは、シミュレーションがすぐに(最初の数ステップで)破綻するか、はじめは長時間実行されるかです。問題が初期コンフォメーション(最小化されていないなど)または系の定義(拘束条件や力など)のいずれかにある場合、通常すぐに破綻します。局所的なエネルギー最小化を実行してみて、それで解決するかどうかみてください。 一方で、ステップサイズが大きすぎる場合、はじめはしばらく実行されることがあります。その場合は、まずステップサイズを小さくしてみてください。
エラーが断続的なものではなく再現性がある場合、通常は何が起こっているのかを正確に追跡することができます。 まず、非常に大きな力がかかっている粒子を探すことから始めてください。そして、その力がどこから来ているのかを特定します。その力は、結合性相互作用によるものか、それとも非結合性相互作用によるものでしょうか? 具体的な相互作用を特定できますか? 例えば、2つの粒子が近すぎるのでは?
これらのエラーをデバッグする別のアプローチは、エラーの原因となるものを系統的に削除することです。力を1つずつ取り除いてみてください。 すべての拘束条件を取り除いてみる。あるいは、周期境界条件を無効にしてみる。少しの辛抱と忍耐で、大抵の場合、原因を特定することができます。
3. 「残基のテンプレートが見つかりません」というエラーが出るのはなぜ?
このエラーは、ForceField上で createSystem() を呼び出したときに発生することがあります。このエラーの意味を理解するためには、OpenMMで力場がどのように動作するかを少し知っておく必要があります。
力場を使って系(System)を構築する最初のステップは、すべての原子のタイプを特定することです。 原子のタイプは非常に固有のものである場合があります。例えば、全ての炭素原子が同じであるわけではありません。力場は、α炭素と芳香族側鎖の炭素とでは、おそらく異なるパラメータを使用するでしょう。 原子タイプは、残基をテンプレートにマッチングさせることで決定されます。力場は、認識できるすべてのタイプの残基のためのテンプレート定義を含んでいます。createSystem()を呼び出すと、渡されたトポロジー(Topology)のすべての残基をループして、それぞれをテンプレートにマッチングさせようとし、そのテンプレートで指定された原子タイプを割り当てます。
残基は2つの情報、すなわち各原子の元素とそれらをつなぐ結合(残基の原子と別の残基の原子をつなぐ結合も含む)の情報に基づいてテンプレートにマッチングされます。残基の名前など他の情報については気にしません。(ただし、後述するように、誤った残基名は問題を引き起こす可能性があります。) 元素と結合だけが重要です。
残基にマッチングするテンプレートを見つけることを妨げる多くの問題があります。ここでは、より一般的なものをいくつか紹介します。
力場のXMLファイルが省略されている。 これは最も単純な可能性の1つです。ForceFieldオブジェクトを作成し、どのファイルを読み込むかを指示したときに、単に1つだけ抜けている可能性があります。例えば、Amber14とCHARMM36の定義では、水を他の力場とは別のファイルに入れ、異なる水モデルを簡単に選択できるようにしています。そのファイルを指定しなかった場合、力場はトポロジー(Topology)の水分子に一致するテンプレートを持ちません。
非標準の残基がある。 AmberやCHARMMのような標準的な力場は、タンパク質や核酸のような様々な標準的な分子に対応しています。しかし、 Topologyにリガンドやその他の低分子が含まれている場合、 力場にはそのためのパラメータがない(つまり、 テンプレートがない)可能性があります。
トポロジーに原子がない。 例えば、 結晶構造には通常、水素原子が含まれておらず、 またフレキシブルな領域では重原子が欠落していることがよくあります。 これらの原子がない場合、トポロジーは力場のテンプレートとマッチングしません。 OpenMM-Setup は、この種の問題の多くを自動的に修正することができます。
水モデルが Topology と矛盾している。 4サイトおよび5サイトの水モデルは、各水分子が仮想サイトのための「余分な粒子」を含んでいることを想定しされています。これがない場合(あるいは逆に、あるのに3サイトの水モデルを使おうとした場合)、テンプレートがマッチングしません。
Modeller.addExtraParticles()を使って、この問題を修正することができます。鎖の終端が正しくない。 タンパク質、そして核酸はさらに、末端の構造が多種多様になっていることがあります。鎖の末端の残基は、中間位置の残基とは違っていて、異なるテンプレートが必要です。多くの場合、力場は各鎖の終端の構造について1つか2つしかサポートしていません。もしTopologyの末端が異なるものである場合、テンプレートにマッチングしません。
非標準のPDBファイルを持っている。 PDBは標準化されたフォーマットであり、specificationで正確に定義されています。 残念ながら、多くのPDBファイルはその仕様に従っておらず、様々な問題を引き起こしています。
最後のポイントについては議論が必要です。PDBファイルには様々な非標準の可能性があるからです。テンプレートは原子の元素と原子間の結合という2つの要素に基づいてマッチングされることを思い出してください。これらの一つでも不正確に再構築されると、テンプレートのマッチングでエラーが生じます。
PDBファイルの各ATOMレコードには、元素を指定するフィールドがあります。残念ながら、そのフィールドが空白のままのファイルもあります。その場合、PDBFileクラスは原子の名前から元素を推測しようとします。通常は正しく推測することができますが、常にそうとは限りません。
結合はより複雑です。PDB の仕様では、標準的な残基と異質原子(heterogen)を区別しています。標準的な残基(アミノ酸、ヌクレオチド、水)には結合が指定されていません。ファイルを読み込むソフトウェアが自動的に結合を追加することになっています。その他の分子はCONECTレコードで結合が明示的に指定されているはずです。これらのレコードがない場合、PDBを読むソフトウェアはどの原子が結合しているのかわからないので、ForceFieldはテンプレートをそれらにマッチングさせることができません。
標準残基には独自の問題があります。それらは名前によって認識されます。 PDB Chemical Components Dictionary ではすべての残基と原子の標準的な名前が指定されています。これにより、例えば、ALAという残基はCAという原子とCBという原子の間に結合があるはずだということがPDBを読むソフトウェアにはわかるわけです。残念ながら、ファイルによっては残基や原子の名前に非標準的なものを使っている場合があります。そのような場合、PDBFileはどこに結合を追加すればよいのかわかりません。
これが、原子や残基の名前が重要になる理由です。ForceFieldはテンプレートをマッチングするときに名前を使いません。しかし、ファイルが非標準的な名前を持っていると、PDBを読むソフトウェアは正しい結合を追加することができず、ForceFieldがテンプレートにマッチングすることができなくなります。
PDBFileは非標準的な名前に対応するように努めています。よく使われる非標準的な名前を認識するための大きなテーブルが用意されています例えば、水分子の標準的な名前は HOH ですが、それ以外にも WAT、SOL、TP3 などの名前も受け付けます。同様に、水分子の酸素の標準的な名前はOですが、OWやOH2という名前も受け付けます。これらの名前は、過去に様々なプログラムが書いたPDB ファイルで誤って使用したもので、OpenMM はそれらを探すことができます。しかし、OpenMMが認識できない他の非標準的な名前を使用している場合、結合は正しく追加されず、ForceField はテンプレートをマッチングさせることができなくなります。
4. SystemやForceに加えた変更が無視されるのはなぜ?
一旦Contextを作成すると、そのContextが作成されたSystemはイミュータブル(不変のオブジェクト)として扱われます。そのシステムに対して更に変更を加えても無視されます。これには、ForcesやVirtualSitesなど、Systemに含まれる他のオブジェクトも含まれます。
Contextの作成にはコストがかかります。GPU カーネルのコンパイル、スプラインの表形式関数へのフィッティング、拘束条件行列の構築と逆行列の計算など、シミュレーションの開始時に行う必要がある先行作業が多くあります。その過程で、Systemからの情報は多くの異なる場所にコピーされ、多くの場合、高度に処理された形でコピーされます。SystemまたはForceに変更が加えられると、多くの内部データ構造が無効になる可能性があります。そのため、OpenMM では、効率と混乱を避けるために、既存のContextはSystemに対するすべての変更を無視するというルールになっています。Contextを作成すると、必要なものはすべてSystemからコピーされ、それ以降は参照されません。
このルールは、状況によっては妨げになる場合があります。シミュレーションの過程で何かを変更することが本当に必要な場合があります。このような状況に対応するために、スピードと柔軟性のトレードオフの関係にある複数の方法が提供されています。
最初の方法はグローバルパラメータです。グローバルパラメータの値はSystemではなくContextに保存されるので、setParameter()を呼び出すことでいつでも変更することができます。カスタマイズしたフォースは全て、グローバルパラメータを定義することができます。 パラメータに依存する形でエネルギーを定義した場合、パラメータの新しい値を設定するだけで、いつでも簡単にエネルギーを変更することができます。 NonbondedForceでは、パラメータのオフセットで同様のことを行うことができます。グローバルパラメータを定義することで、粒子のセットを一度に協調的に変更することができます。
次に、ほとんどのフォースではupdateParametersInContext()メソッドが用意されています。これはSystemの限られた部分を更新するための制御された方法を提供します。例えば、粒子のセットの電荷を変更することができます。これを行うには、NonbondedForceに対してsetParticleParameters()を呼び出します。 他の変更と同様に、これは既存のContextでは無視されます。 しかし、次にupdateParametersInContext() を呼び出すと、変更されたパラメータ値が既存のContextにコピーされます。これは、グローバルなパラメータを変更するだけの方法よりもはるかに遅いですが、完全に新しいContextを作成するよりもはるかに速いです。この方法では、Forceのすべての条件を変更できるわけではないことに注意しましょう。updateParametersInContext() メソッドが何を更新し、何を更新しないかについては、各メソッドの API ドキュメントを参照してください。
最後の選択肢は、Context上でreinitialize()を呼び出すことです。 これにより、内部のデータ構造をすべて破棄し、一から作り直します。 これは、事実上まったく新しい Context を作成するのと同じことです。非常に遅いですが、絶対に全てを変更することができます。
内部データ構造がすべて破棄されるため、位置、速度、パラメータ値などの状態の情報も失われます。引数 reinitialize(preserveState=True)を渡すことで、それらの情報を保持するように要求することができます。これを実行すると、まずメモリ上にチェックポイントを作成し、次に Context を再初期化(reinitialize)し、最後にチェックポイントを再読み込みします。ほとんどの変更に対して、これは簡単に状態の情報を保持する方法を提供しますが、もしチェックポイントを有効でなくするような方法でSystemが変更された場合は、失敗して例外を発生させます。例えば、System内の粒子の数が変化した場合、チェックポイントをロードすることができなくなり、例外が発生し、状態の情報が失われます。
以上が、2022年4月現在のFAQです。
OpenMMをステップバイステップで 〜 Part 4 ColabでOpenMM シミュレーションを実行 〜
OpenMMの使い方を順番にたどる記事のPart 4です。過去の記事で、タンパク質構造の前処理(Part 1、Part 2)とシミュレーション実行のためのスクリプト(Part 3)を手に入れるところまでが終わりました。
あとは計算を実行するだけ!でも大変そうだからColabでやってしまおう!ってなお話です。*1
1. 準備
1-1. 使用ファイル
今回使用するファイルは以下の2つです。
- ① PDB ID: 3POZをMDシミュレーション用に処理したファイル(
3poz-processed.pdb) - ② OpenMMシミュレーションを実行するスクリプト(
run_openmm_simulation.py)
それぞれの作成手順は過去の記事をご覧いただければと思います。
処理済みのPDBファイルはこんな見た目です。
1-2. スクリプトの修正
Google Colabでシミュレーションを実行する前に、前回作成したスクリプトを少し修正しました。
修正内容は2点です。
- ① シミュレーションの途中経過(残り時間 etc.)をColabノートブック上に表示させる
- ② シミュレーション後の最終状態をPDBx/mmCIFフォーマットでも出力する
1-2-1. 途中経過の出力
MDシミュレーションのトラジェクトリ生成段階(プロダクション)は計算時間がかかるので、「計算そのものが進んでいるか?」「どれくらい時間がかかるのか?」心配になります。
計算のログ自体はログファイル(log.txt)に書き出すようにスクリプトで設定されていますが、計算途中でも確認できた方が安心できそうです。
そこでColabノートブックのセルの出力にも同様の内容を書き出すことにしました。

上図のように、プロダクションを行う「simulation.step(steps)」の直前にもう一つStateDataReporter()を追加しました。標準出力(stdout)に進捗情報を表示するためのコードです。
from sys import stdout simulation.reporters.append(StateDataReporter(stdout, 1000, step=True, progress=True, remainingTime=True, speed=True, totalSteps=steps, separator="\t"))
1-2-2. 最終状態のPDBx/mmCIF出力
前回のスクリプト作成では、シミュレーション後の出力ファイルをXMLにしていました。
# Write file with final simulation state simulation.saveState("final_state.xml")
最終状態の系を可視化するならPyMolで読み込めた方が楽だよね!と、いうことで構造を出力するようにしました。

OpenMM SetupのOutputタブで「Save final simulation state:PDBx/mmCIF (no velocities)」とすると上記のようになります。
この状態のスクリプトを使っても良いですし、シミュレーション終了後に同じコードを実行してもファイルを書き出すことができます。
state = simulation.context.getState(getPositions=True, enforcePeriodicBox=system.usesPeriodicBoundaryConditions()) with open("final_state.pdbx", mode="w") as file: PDBxFile.writeFile(simulation.topology, state.getPositions(), file)
2. Google Colabでシミュレーション
2-1. 手順
それではGoogle Colabに計算してもらいましょう!
手順は以下です。*2

くどくど書いてますが、インストールしてスクリプト(.py)を実行するだけです。
2-2. 実行
では実際に計算を進めていきましょう!
2-2-1. GPUに設定
Colabにログインして新しいノートブックをつくったら忘れずにハードウェアをGPUにしておきます。
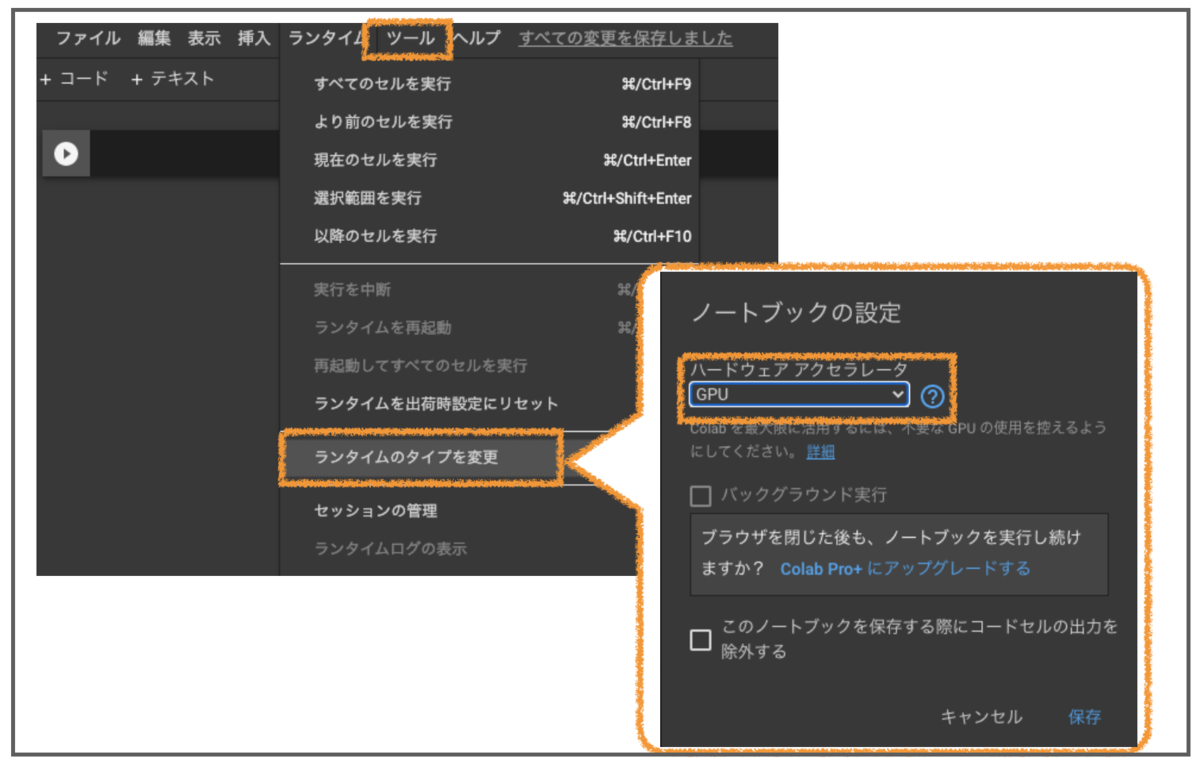
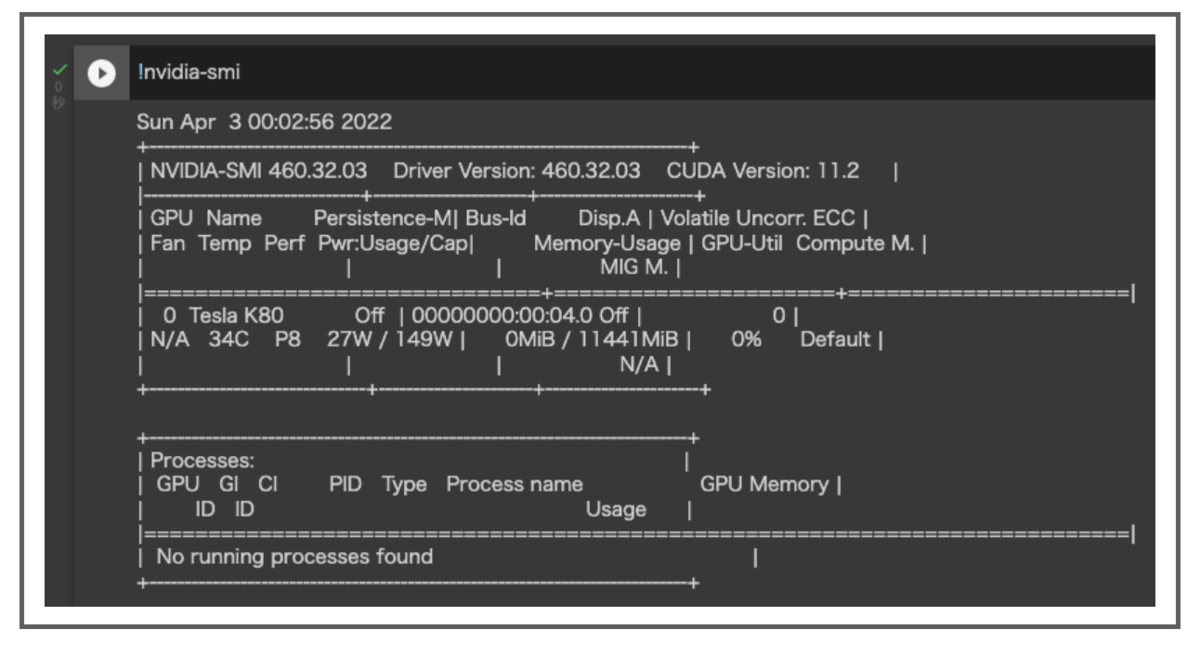
以下をセルで実行して、GPUに切り替わったことを確認することもできます。
!nvidia-smi
2-2-2. OpenMMのインストール
次に、OpenMMをインストールします。
OpenMMはcondaがあれば簡単にインストールできるので、最初にminicondaをインストールします。
!wget -qnc https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh !bash Miniconda3-latest-Linux-x86_64.sh -bfp /usr/local !rm Miniconda3-latest-Linux-x86_64.sh
wgetでLinux用のminicondaを取ってきて、「/usr/local」にインストールしています。終わったら不要なファイルをrmで削除してます。
次に、OpenMMをconda-forgeチャンネルからインストールします。
!conda install -c conda-forge openmm=7.6 python=3.7 -y
インストールできたらOpenMMが使えるようにパスを通しておきます。
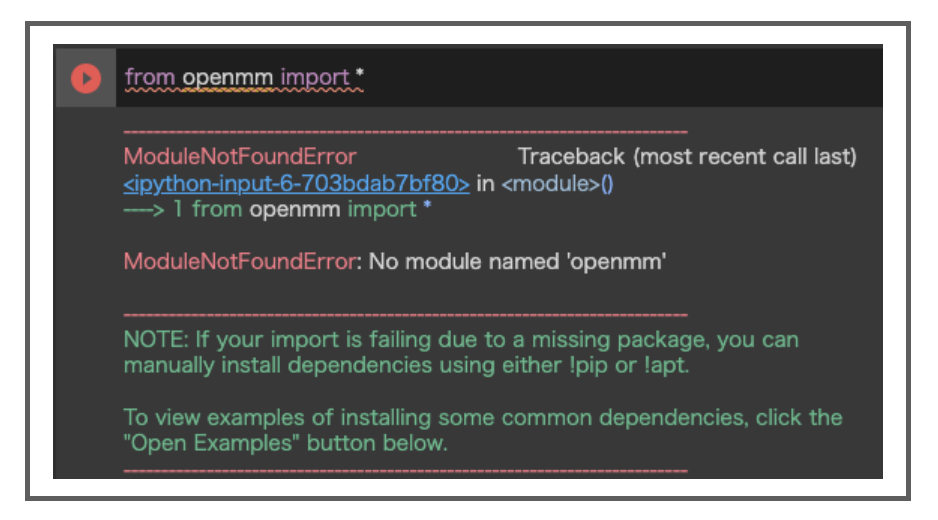
import sys sys.path.append('/usr/local/lib/python3.7/site-packages/')
忘れててModuleNotFoundErrorで怒られました。
2-2-3. ファイルアップロード
Colabに計算で使用するPDBファイル(3poz-processed.pdb)とスクリプト(run_openmm_simulation.py)をアップロードします。
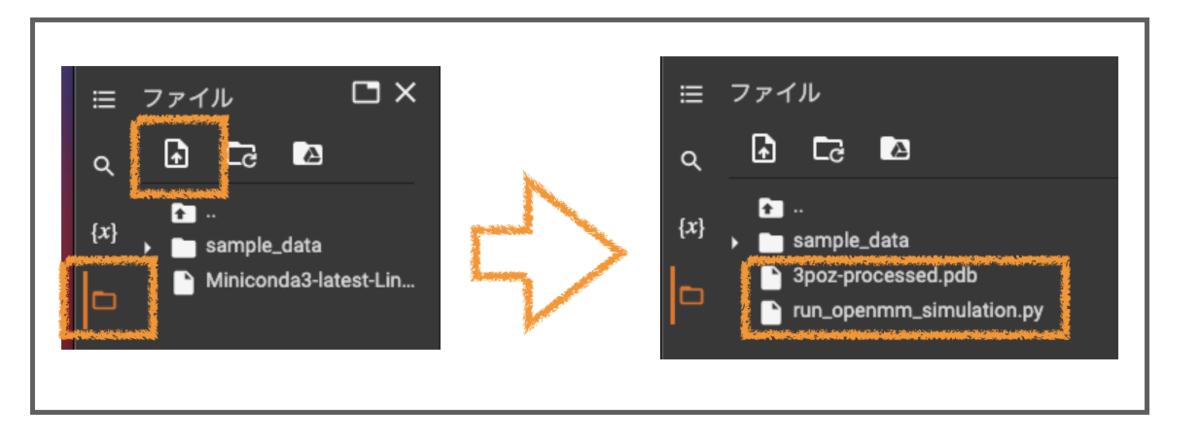
Colabの左側でクリックするだけ。前からこんなに簡単でしたっけ?
2-2-4. スクリプトの実行
あとはスクリプトを実行するだけ!
%run run_openmm_simulation.py
標準出力に経過を出すように書き換えたので進捗が確認できます。
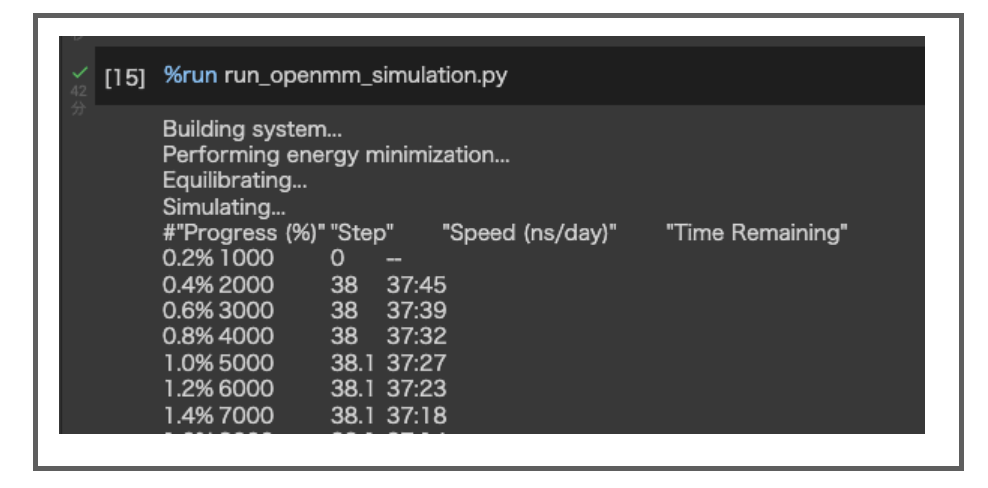
今回はトラジェクトリの長さを1nsとしていますが、プロダクションだけでおおよそ40分程度かかりました(平衡化などを含めると小一時間くらい)。
2-2-4. 結果のダウンロード
計算が終わると左側ファイルに色々と新しいファイルができています。
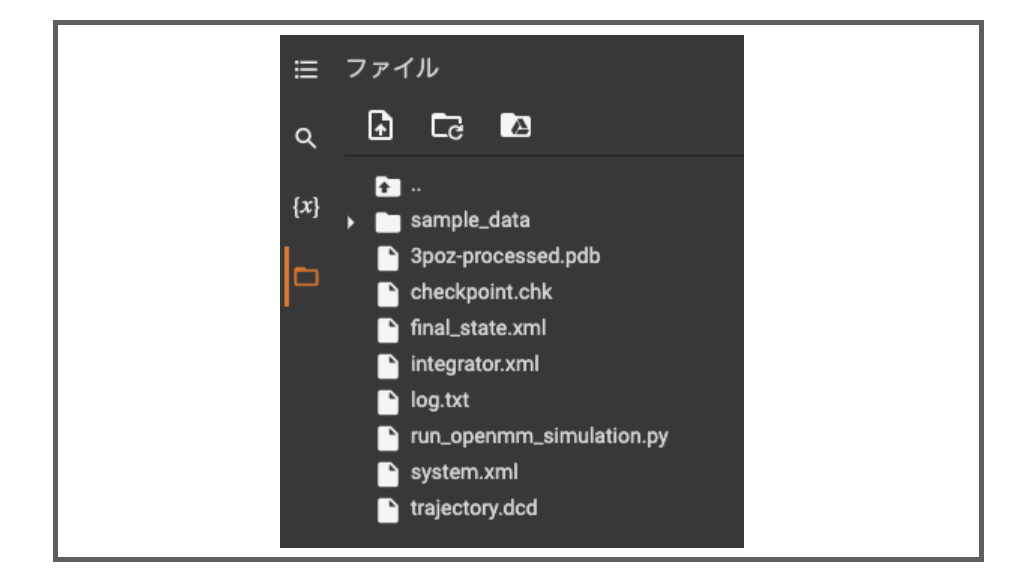
google.colabのfilesを使って、簡単にローカルPCにダウンロードできます。*3
from google.colab import files files.download("trajectory.dcd") files.download("log.txt") files.download("checkpoint.chk") files.download("system.xml") files.download("final_state.xml") files.download("final_state.pdbx")
Colabはランタイムが切断されるとファイルが全て消えてしまうので、とりあえず目につくものをダウンロードしておきました。
容量が大きいファイルはブラウザが「本当にダウンロードして良いの?」と聞いてくる場合があるので許可してください。*4
PCではなくGoogle Driveに保存したい時はマウントしてコピーすれば良いそうです。
例えば以下でGooge Driveのマイドライブディレクトリに「trajectory.dcd」が保存されます。
from google.colab import drive drive.mount('/content/drive') !cp trajectory.dcd /content/drive/MyDrive/trajectory.dcd
Colabの左側に出ているデフォルトのディレクトリ名はcontentとなっているようです。上記でcontent/driveにGoogle Driveがマウントされます。
パスに困った時は左側に表示されるディレクトリで「パスをコピー」すればOKです。

3. PyMolで可視化
結果のファイルが手に入りました!取り急ぎPyMolで可視化してみましょう。
シミュレーションの最終時点のファイルは「final_state.pdbx」です。OpenMM Setupのデフォルトのファイル名では拡張子が「pdbx」となっているので「.cif」に名前を変えてからPyMolに入れました。

境界からタンパク質がはみ出したぞ????なんでや???
よくわからないですが動画にしました。構造を読んだ状態でトラジェクトリ(trajectory.dcd)をPyMolに読み込ませれば良さそうです。
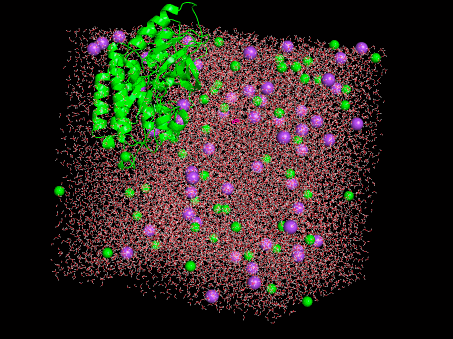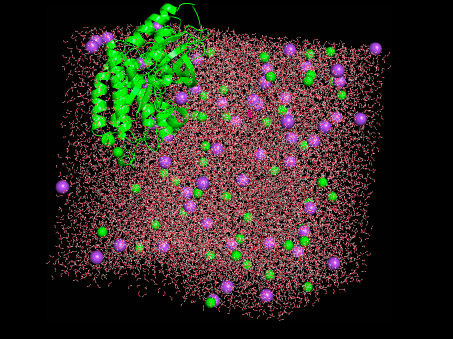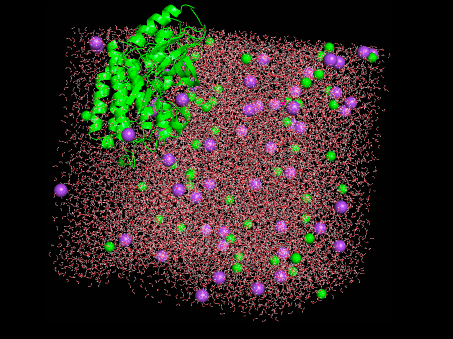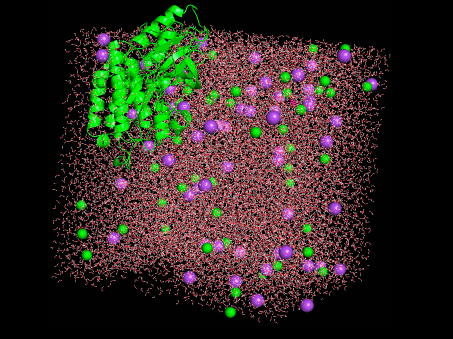
かわいい!!・・・けど、はみ出してる。。。
4. Center of Mass Motion?
境界からタンパク質が出てしまっている原因がよくわからないので検索するとCenter of Mass Motionという用語に行き当たりました。OpenMM User Guideに3.6.12 Removing Center of Mass Motionという項目があります。
曰く、デフォルトではOpenMMアプリケーションは「CMMotionRemover」を系に付与しており、「各ステップ毎に重心の動きをすべて取り除くことで、時間発展に伴い系全体がドリフトしてしまうことを防ぐ」のだそう、、、。
ドリフトが何かよく分かりませんが、タンパク質が境界外に出るのはかなり「drift(漂流)」感のある動きです。
上記項目では、「removeCMMotion=Falseを設定すればドリフトを許容することもできるけど、かなりレアなシミュレーションだよねー」と書いてありました。
デフォルトから変えていないので重心は動かない設定のはずなのですが・・・。心当たりのある方、ご教示いただけると嬉しいです。
5. おわりに
以上、今回はGoogle Colab上でOpenMMの計算を実行する方法について見てみました。手順がくどくて読みにくい記事になってしまいましたが、記憶力の悪い私の備忘録ということでお許しください。
得られた結果を可視化するとタンパク質が境界外に出ているというなんとも気持ちが悪いものでした。何はともあれ、一応は動きのある結果が得られたということで、Colab上での計算手順は確認できたのでヨシ!
・・・困った。
以上、今回もかなり間違いが多そうです。お詳しい方ご教示いただけると嬉しいです。
ではでは!
6. 追記
境界からタンパク質がはみ出ることについてよくわからないと書きましたが、OpenMMのGitHub Frequently Asked Questionsにそのものズバリ書いてありました。
「Why does my protein stick out past the edge of the water box?」
「問題ないよ!周期境界条件(periodic boundary conditions)をきちんと理解していないだけだよ!」ってな感じみたいです。
シミュレーション本番においても、計算後のトラジェクトリ描画においても、「境界をどのようにとるか?」「分子をどのように描画するか?」によって見え方が異なってくるようです。
GitHubのIssue #2688 Simple example of using SystemBuilder to simulate protein-ligand complexの議論中でも、タンパク質が境界から飛び出た図が引用されていますが、特におかしい挙動ではない(The trajectory produced looks just fine)と書かれています。
以上、同様の描画が得られて気になる方はご参照いただければと思います。
*1:イラストはいらすとやさんより
*2:Making it rainの手順を参考にさせていただいています。
*3:参考:Google Colabのファイルのアップロードとダウンロード
*4:ダウンロードが始まらないと思ってたら、ポップアップが出てました。気づかなかった。。。
OpenMMをステップバイステップで 〜 Part 3:GUIでシミュレーション条件の設定 〜
OpenMMの使い方を順番にたどる記事のPart 3です。Part 1、Part 2の記事でMD計算のためのタンパク質構造の前処理が完了しました。今回の記事ではシミュレーション条件本体のセットアップの流れを追ってみたいと思います。
利用するツールはOpenMMのGUI、OpenMM Setupです。なお、今回の記事では設定のみで計算そのものは実行しません。*1
1. OpenMM Setupでできること
まず最初にOpenMM Setupでできることの概要です。
前々回の記事でPDBファイルを読み込んで構造の前処理の方法を確認しました。この時はPDBの出力で一度終わりとしましたが、そのまま続けてシミュレーション条件を指定するスクリプト(run_openmm_simulation.py)の作成と保存、さらに実行(Run Simulation)までを行うことができます。
画面構成はこんな感じ。

上図のように、左側で選んだ条件が右側のスクリプトに逐次的に反映されて設定ファイルが出来上がっていきます。
前処理と同じく指示に従って条件を選択していくだけ!これなら私でもできそう!
2. MD計算の流れと設定項目のおさらい
シミュレーション条件を設定していく前に、MD計算の流れと設定項目のおさらいをしておきましょう。
計算の流れはざっと以下です。
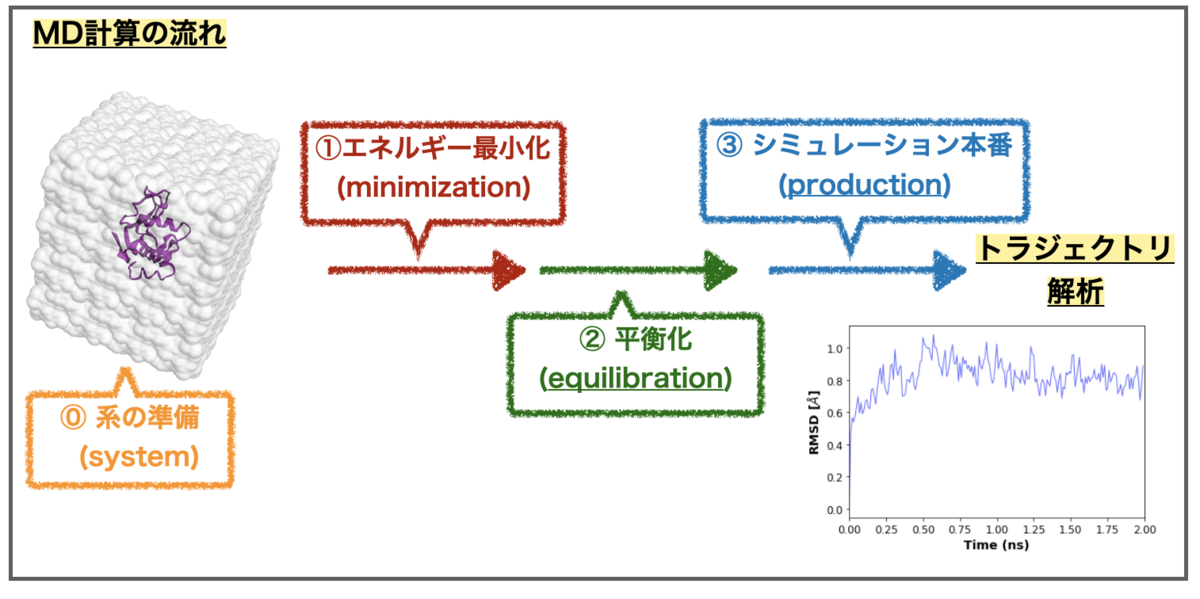
準備直後の系(⓪)は水分子やイオンを適当に配置しただけなので、最初にエネルギー最小化(①)します。ついで各原子にランダムに速度を与え、徐々に系全体を設定温度まで引き上げていきます(平衡化(②))。最後に平衡状態の系においてアンサンブルを達成するようにトラジェクトリを生成します(シミュレーション本番(③))。*2
設定の必要な項目は大体こんな感じ。*3

ごちゃごちゃしてますが、系の初期状態と平衡状態(のアンサンブル)を定めて、そこに至る計算方法(分子力場、インテグレータ)や、状態の制御方法(サーモスタット、バロスタット)を設定します。あとは希望のシミュレーション時間(ステップ数)計算すれば良さそうです。
また、MD計算はヘビーなので計算環境(GPUをつかえるか?)やログ、結果(トラジェクトリ)の出力管理といった項目も大事な設定となってくるようです。
これからOpenMM Setupの4つのタブで順番に設定を確認していきますが、おおよそ以下の対応になっています。

では順番に見ていきましょう!
3. OpenMM Setupでシミュレーション条件を設定しよう!
3-1. 注意!~ 入力手順とデフォルト値の違い
設定を始めていく前に少し注意点があります。OpenMM Setupでは入力の手順で少し表示内容が変わります。
具体的にはPDBファイルを入力とした時、「① 未処理の構造を入力としてOpenMM SetupでClean upしたあと、続けてシミュレーションの設定に進んだ場合(Part 1 記事でご紹介した手順)」と、「② すでに処理済みのPDBファイルを入力として直接シミュレーションの設定に進んだ場合」です。

上図左のように前者(①:Yes. Let's clean it up now.)では、シミュレーションの設定項目のほとんどが空欄でひとつずつ自分で選んでいきます。一方、後者(②:No. my file is all ready to simulate.)ではデフォルトの設定(数値)が最初から記入されていて、変更したい箇所だけを変えればOKです。
Clean upをした後でデフォルトの数値設定が知りたい場合は、処理後のPDBファイルを一度保存して、再度読み込んで最初からはじめればOKです。
3-2. Systemタブ
それではひとつずつタブを見ていきましょう。まずはSystemタブです。
「System(系)」とついていますが、ここでは主に長距離相互作用(静電相互作用)の計算方法を設定します。
先の項(3-1.)で見た通り、タンパク質構造の前処理からそのまま進んだ時は項目の大半が空欄です。Systemでは以下のようになっています。

Nonbonded MethodやConstraintsといった項目があります。右側のスクリプトでは「# System Configuration 以下」に対応していることがわかります。
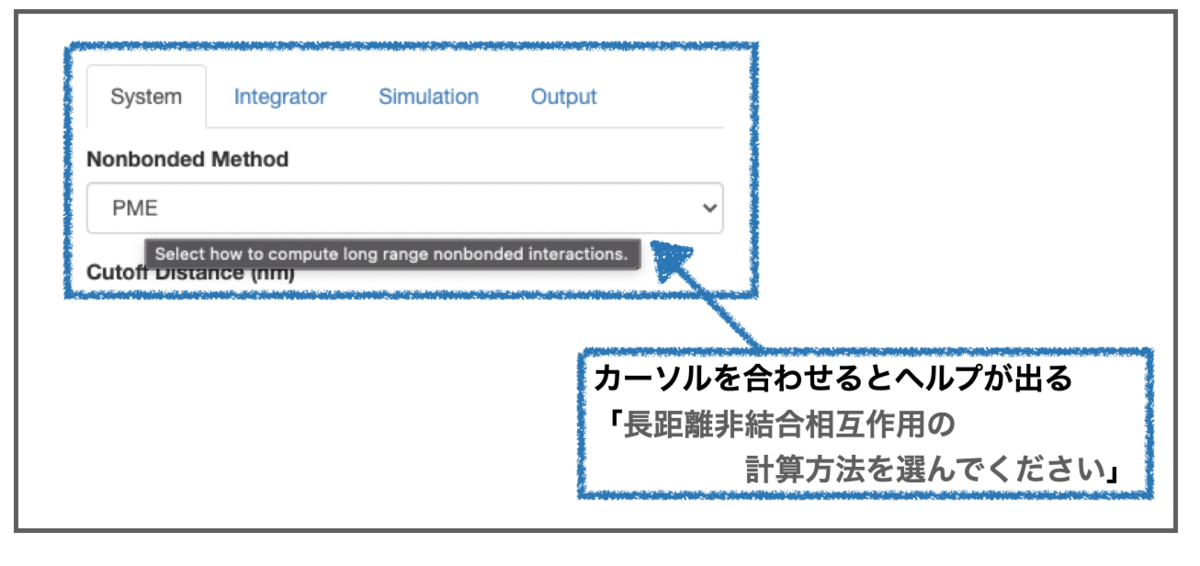
左側の各欄にカーソルを合わせると「何を設定する項目か?」ヘルプが表示されます。
今回はTeachOpenCADDのT019 · Molecular dynamics simulationやデフォルト値を参考に以下のように設定してみました。

まず、① 長距離静電相互作用(nonbondedMethod)はPME(粒子・メッシュ・エバルト法、エバルトの方法 - Wikipedia)です。*4
② カットオフ距離(nonbondedCutoff)はPME法のうち、「粒子部分(実空間における短距離ポテンシャルの直接和)」に関するパラメータで、「直接計算する距離をどの程度の長さにするか?」を定めるカットオフ値のことだと思います。*5
③ Ewald法の誤差許容範囲(ewaldErrorTolerance)は、「Ewald法で和を取る際に、切り捨て計算することで生じる力の端数の誤差」に、おおよそ等しくなるように設定します。デフォルトでは0.0005です。*6
④ 拘束条件(constraints)では、結合長や結合角に関する拘束をオプションで設定することができます。HBondsとした場合は、「水素原子を含む全ての結合の結合長」を拘束します。他にはAllBondsやHAnglesといった選択肢があります。*7
⑤ 拘束条件の誤差許容範囲(constraintTolerance)は、「拘束条件について誤差をどの程度許容するか?」で、1x10-6としています。*8
なお、スクリプト側にある「⑥ 剛体としての水(ridigdWater)」は水分子の扱いを決めるパラメータです。OpenMMでは水分子を結合長・結合角ともに拘束された、完全に剛直なもの(rigid)として扱います。Falseにするとflexible waterとなります。
以上がSystemタブでした。*9
3-3. Integratorタブ
つづいてIntegratorタブです。積分計算やアンサンブルについて設定します。
こんな感じで設定してみました。
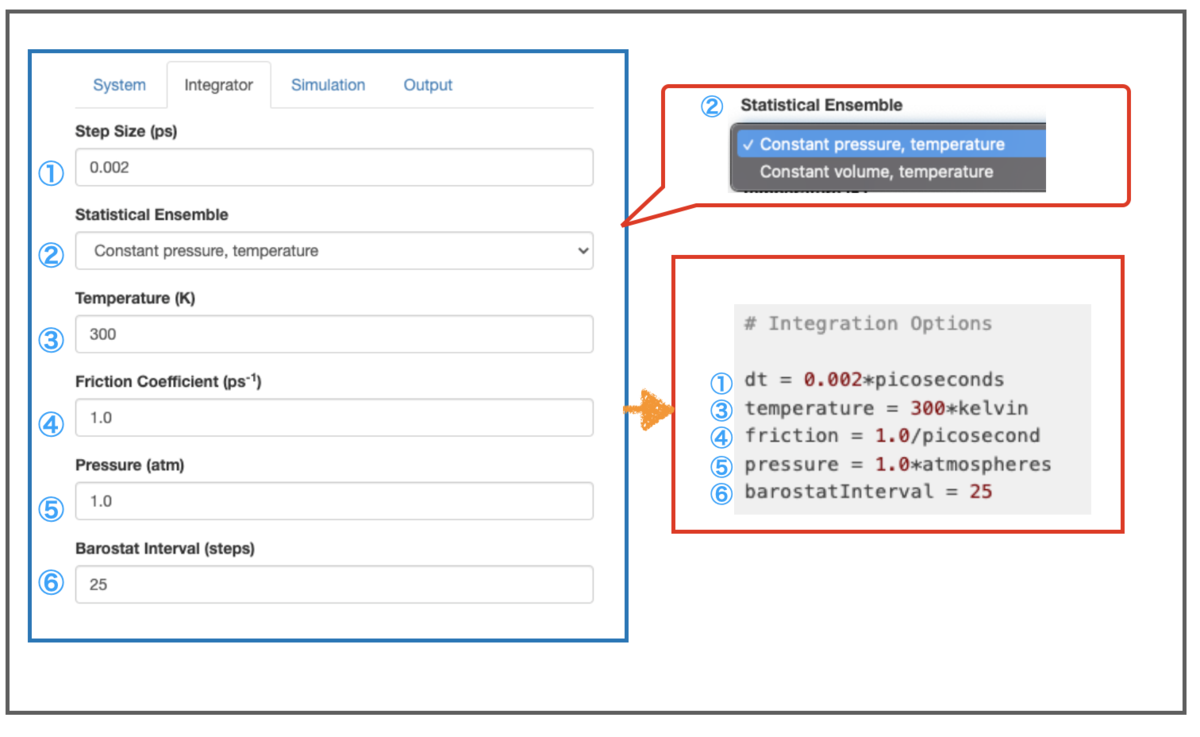
まず、① 積分計算の時間間隔(Step Size, dt)は「0.002ピコ秒(2フェムト秒)」としました。
② 統計的アンサンブル(Statistical Ensemble)には、「Constant pressure, temperature, NPT」と「Constant volume, temperature, NVT」の選択肢があります。今回はNPTとしました。
尚、GUIのOpenMM Setupでは「アンサンブルを達成するための計算方法(温度や圧力を一定に保つための計算方法)」を選択する項目は無いようです。NPTを選択するとデフォルトで「温度:LangevinMiddleIntegrator、圧力:MonteCarloBarostat」が設定され、スクリプトに反映されます。
③ 温度(temperature)は「300K(27℃)」にしました。
④ 摩擦係数(Friction Coefficient, friction)は温度制御に関わるパラメータです。デフォルトのLangevinMiddleIntegratorはランジュバン動力学に基づくものなので、ランジュバン方程式の摩擦項の係数をパラメータとして設定します。*10*11
⑤ 圧力(pressure)の単位は「atm(標準大気圧)」です。
⑥ 圧力調整の間隔(Barostat Interval, barostatInterval)は、「何ステップごとに圧力調整を行うか?」です。①で「Step size : 0.002 ps」としているので、⑥で間隔を「25 steps」とすると「0.002 x 25 = 0.050 ps」毎に調整を行います。*12*13
以上がIntegratorタブでした。
なお、上図のスクリプト部分は変数の設定のみです。計算方法の設定は以下のようになっていました。
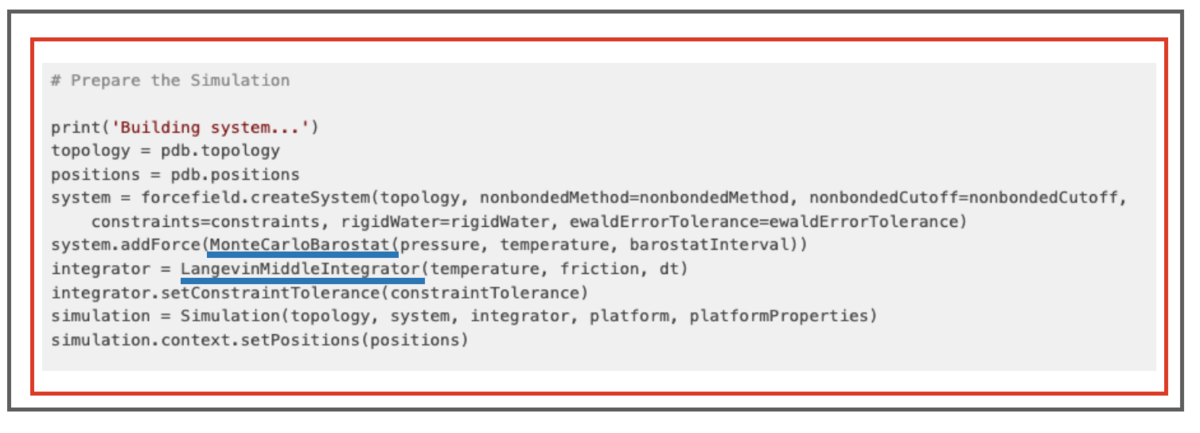
積分が LangevinMiddleIntegratorや圧調整が MonteCarloBarostat となっていることが確認できますね。
3-4. Simulationタブ
続いてSimulationタブです。シミュレーション時間や計算環境の設定を行います。
Google ColabのGPUを利用するつもりで、以下のように設定してみました。

① シミュレーションの長さはトラジェクトリを生成する時間(production)で、単位はstepです。
Integratorタブで積分のStep Sizeを「0.002 ps」としました。従って、上図のように「500,000 steps」とすると、「0.002 x 500,000 = 1,000 ps = 1 ns」となり、1 ナノ秒のシミュレーションとなります。
② 平衡化の長さも同様にstepで長さを指定します。上図では「50,000 steps」(0.1 ナノ秒)としました。
尚、上記の時間設定はかなり適当なので参考にしないでください。目的にあった適切な値を選択してください。*14
③ プラットフォームでは計算環境を設定します。選択肢にはReference、CPU、CUDA、OpenCLがあります。Google ColabではGPUを使用できるのでCUDAとしています。*15
④ 精度はプラットフォームでCUDAあるいはOpenCLを選択した場合に選べるパラメータで、「single、mixed、double」があります。
精度よりも速度を重視するなら「single(単精度)」で、逆に精度を重視するなら「double(倍精度)」です。「mixed」は力をsingle、積分をdoubleで計算する混合モードです。*16
以上がSimulationタブでした。
3-5. Outputタブ
最後にOutputタブです。結果の出力を設定します。色々な形式で出力できて、項目数が多いので分割して見ていきます。
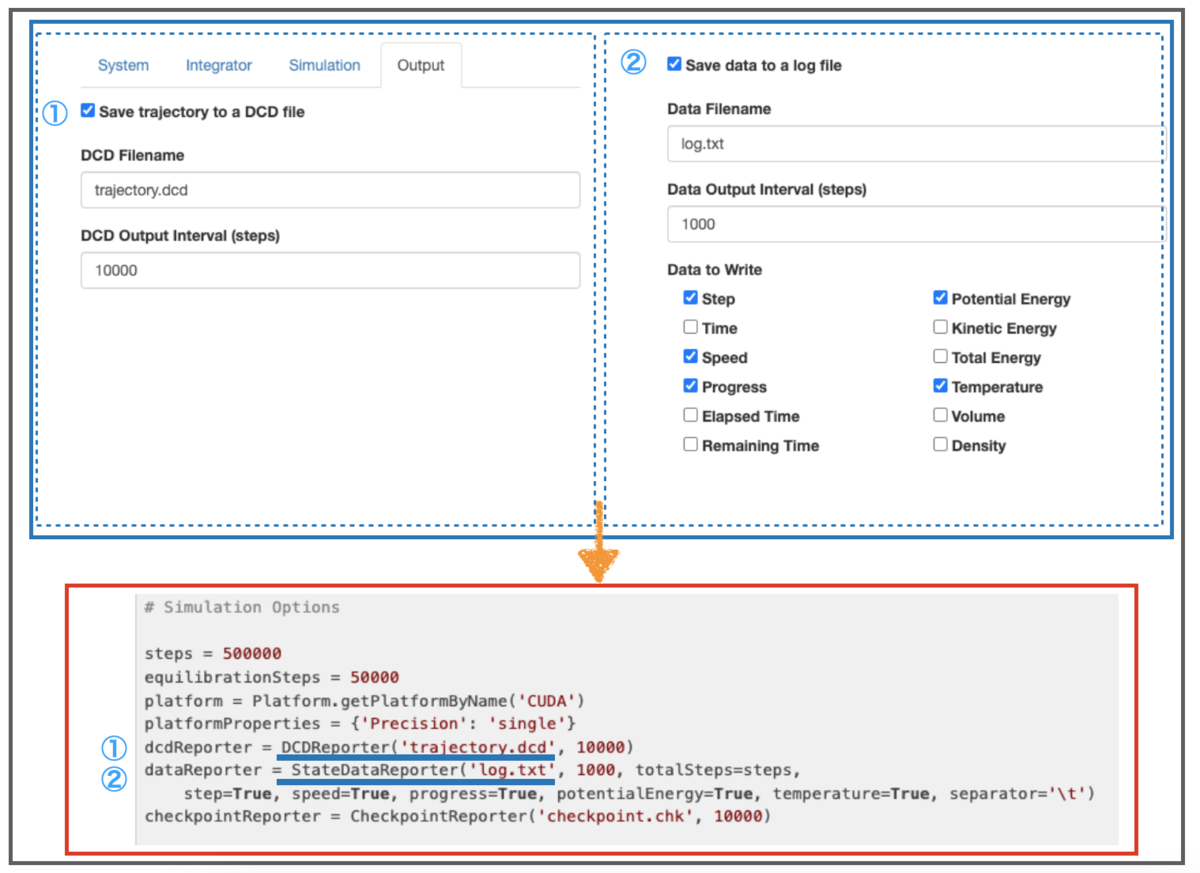
OpenMMではトラジェクトリをPDB、PDBx/mmCIFあるいはDCDフォーマットで保存することができます。*17GUIのOpenMM Setupでは、このうちDCDフォーマットのみを選択できます。*18
まず、① ボックスにチェックを入れて「トラジェクトリを保存する」を選択し、ファイル名と「どれくらいの間隔(step)ごとに結果を出力するか?」を指定します。
上図では「trajectory.dcd」ファイルに「10000 steps (20 ピコ秒)」毎に状態を書き込むことになります。
また、計算のログファイルを残すこともできます(②)。プロダクションタイムのトラジェクトリ以外のさまざまなデータを保存することができます。
保存する内容はData to Writeの中から選べ、エネルギーや速度、温度などがあります。上記では、「log.txt」ファイルに「1000 steps(2 ピコ秒)」毎に書き込みます。*19
①、②で設定した内容はスクリプトではそれぞれ①DCDReporter( )、②StateDataReporter( )に引数として反映されています。

OpenMMではシミュレーションの進行具合をチェックポイントファイルというバイナリファイル(.chk)で残すことができます(③)。サーバーエラーなどでシミュレーションが途中で止まってしまった時に、チェックポイントファイルを読み込んで計算を再開することができるそうです。
上図では、「checkpoint.chk」ファイルに「10000 steps (20 ピコ秒)」毎に上書き保存しています。スクリプトでは③CheckpointReporter( )です。*20
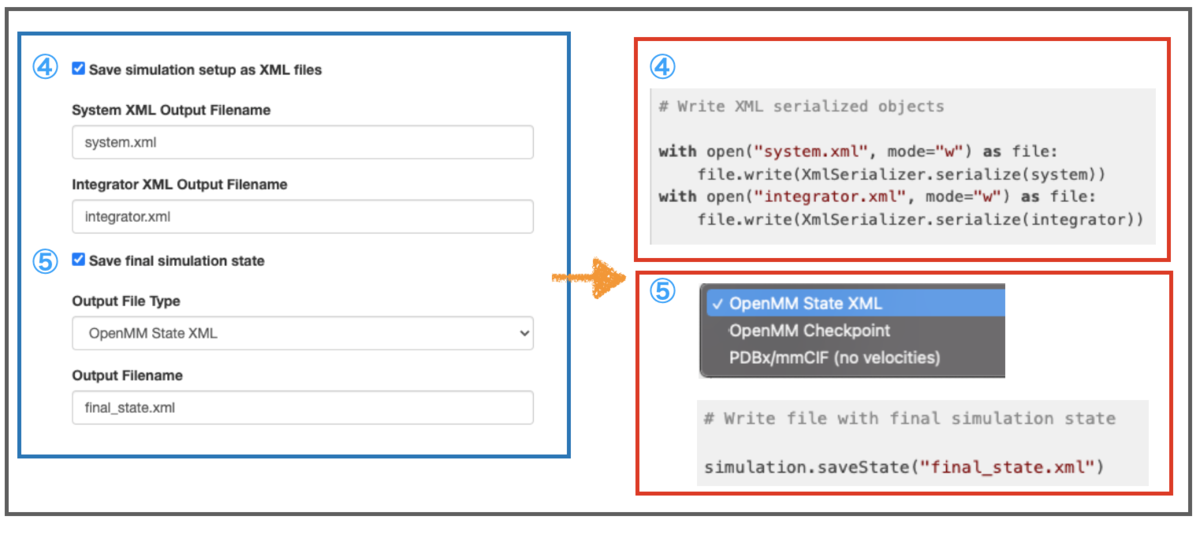
また、シミュレーションの状態(位置、速度 etc.)をXMLファイルに書き出すこともできます。このXMLファイルを再度読み込むことでシミュレーションを続けることができます。*21
chkには「同じ計算環境、OpenMMのバージョンでしかシミュレーションを再開できない」という欠点がありますが、XMLにはその欠点がありません。一方で、XMLにはサイズが大きくなるという欠点があります。
上図では、シミュレーションの設定(④)と、最後の状態(⑤)をそれぞれXMLで保存しています。
スクリプトでは、④シミュレーションの設定「system.xml、integrator.xml」はテキストファイルに書き込む形式で書かれており、⑤最後の「final_state.xml」はsaveState( )というメソッドを使った形式になっています。
なお、⑤はXML以外に、chkやPDBx/mmCIFフォーマットを選択することができます。
以上がOutputタブでした。
3-6. スクリプトを保存
これで4つのタブでの設定がすべて終わりました。最後に作成されたスクリプトを保存しておきましょう。

Save Scriptをクリックすると「run_openmm_simulation.py」というファイルが保存されます。
4. おわりに
以上、今回はOpenMM Setupを使ったシミュレーション条件の設定をみてみました。GUIでクリックしていくことで逐次的にスクリプトが生成され、同時に確認できるというのはとても分かりやすくて良いですね!
以前の記事と合わせて、タンパク質構造とシミュレーションスクリプトが揃ったことになります。あとはこれが本当に動くのかどうかが心配です・・・。 早速試したいところですが、記事が長くなってしまったので続きは次回に。
記事の途中、設定の説明を試みていますがOpenMM User Guideを適当に訳しただけなので、意味のわからないところ、間違っているところが多いと思います。ご指摘いただけると嬉しいです。
おしまい。
*1:今回の記事はOpenMM User Guideの3. Runnning Simulationの記述を参考にしています。
*2:参考:MDシミュレーションのチュートリアル〜PDB: 1LKEの場合〜
*3:イラストはいらすとやさんより
*4:プルダウンで選べるPME以外の選択肢は図の右上
*5:OpenMM User Guide 3.1. A First Exampleでは「cutoff for direct space interactions」となっています。湯どうふさんのYouTubeの解説動画「分子シミュレーション実行時のクーロンポテンシャルの効率的な計算法:Ewald法とParticle Mesh Ewald法【理論化学、計算科学】」がとても分かりやすいです。
*6:OpenMM User Guide 3.6.5. Nonbonded Interactions
*7:OpenMM User Guide 3.6.6. Constraints
*8:OpneMMのGitHub Constraint Tolerance Guidelinesでは「1x10-5と1x10-6のどちらが良いか?」ディスカッションされています。「温度一定なら1x10-5、エネルギー一定なら1x10-6」といった意見が出ています。
*9:今回は設定しませんでしたがHydogen Mass Repartitioningは「重原子に結合する水素原子の重さを変えて、動きの速い水素原子を遅くする」ものだそうです。OpenMM User Guide 3.6.7. Heacy Hydrogens
*11:OpenMM User Guide 3.6.8. Integrators
*12:OpenMM User Guide 3.6.10. Pressure Coupling
*13:MDの圧力調整におけるモンテカルロ法(Monte Carlo barostat)については、森野キートスさんのブログ記事「AmberでMD計算するときの圧力制御法の違い」などが参考になります。
*14:OpenMM Setupのデフォルト値では、「Step Size :0.004 ps、Simulation Length :1000000 steps、Equilibration Length:1000 steps」のようです。
*15:それぞれの選択肢の詳細はOpenMM User Guide 7.7. Platformsをご参照ください。Referenceはパフォーマンスは考えずに読みやすいコードにしたもので、他の選択肢はそれぞれのプラットフォームに合わせてパフォーマンスを最適化しています。
*16:各プラットフォームで選択できる精度を含む個別のパラメータについてはOpenMM User Guide 10. Platform-Specific Propertiesをご参照ください。
*17:OpenMM User Guide 3.6.13. Writing Trajectories
*18:DCDはMD計算のトラジェクトリでよく使われるフォーマットのようです。参考:DCD Plugin, Version 1.11
*19:OpenMM User Guide 3.6.14. Recording Other Data
*20:OpenMM User Guide 3.6.15. Saving Simulation Progress and Results
*21:OpenMM User Guide 3.6.15. Saving Simulation Progress and Results