MD計算のためのオープンソースソフトウェアをさらっと
前回、前々回の記事でMaking-it-rainというプロジェクトを利用して分子動力学(MD)計算をGoogleColab上で実行できることと、そのパラメータについてお勉強してみました。
計算科学の分野はオープンサイエンスの流れが進んでいて、MD計算に関連するオープンソースソフトウェア(OSS)も色々とあるようです。Making-it-rainもOSSをベースに構成されています。
せっかく遊ぶならどうやって動いているのかも知りたいよね!
というわけで(?)、Makig-it-rain(とついでにTeachOpenCADD)を参考に、MD計算に関するオープンソースツールにどんなものがあるか?さらっと眺めてみました!

1. 取り上げるツール
この記事ではMaking it rainとTeachOpenCADD (T19, T20)で出てくるツールをコピペしていきます。参考資料の都合上、ここでとりあげるツールはタンパク質(+リガンドの複合体)のMD計算に偏っていそうです。ご留意ください。
こんな感じのツールが出てきました。
- MD計算本体を実行するためのツール
- 分子力場(低分子用?)
- 計算対象の前処理やファイルの変換
- 結果(トラジェクトリ)の解析
いっぱいだ!
2. MD計算OSSの本命? OpenMM
2-1. 概要
まずはOpenMMです。これが本丸(?)のソフトみたいなので、最初からクライマックス。
有名なソフトらしく日本語で検索してもWikipediaの記事(OpenMM - Wikipedia)や、使い方を解説してくださっている記事がヒットします。
OpenMMは、分子動力学シミュレーションのためのオープンソースのライブラリであり、パンデ研究室によって管理されている。Python、C++、Fortranなどの言語に対応しており、GPUで高速化する機能を持っている。また、AMOEBA、GROMACS、AMBERなどの他の分子シミュレーションツールとのインタフェースも可能である。スタンフォード大学の研究者ビジェイ・S・パンデが作成したこのプロジェクトは、GitHubで自由に利用できるようになっている。(Wikipedia - OpenMMより引用)
こんな感じ。
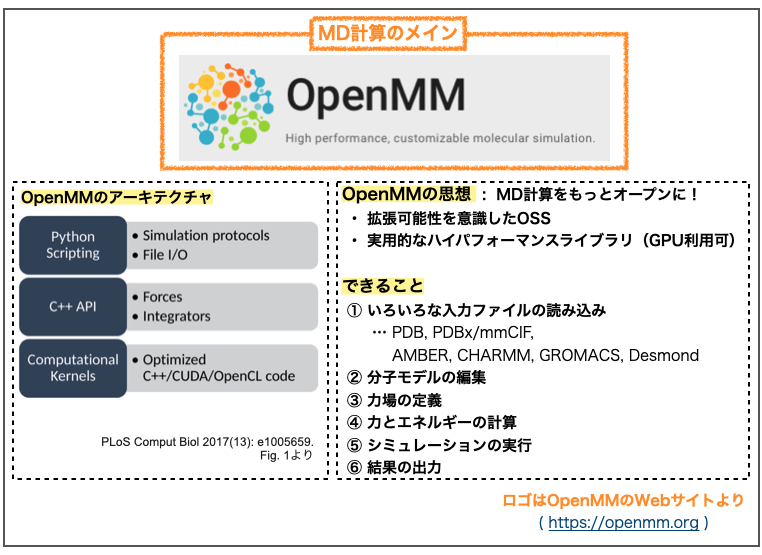
基本的にMD計算に必要なものが揃って提供されているようですね!ファイルの読み込み(①)から、シミュレーションの系の設定(②, ③)と計算の実行(④, ⑤)、結果の出力(⑥)までできるそうです。
2022年現在のバージョンはOpenMM 7.7で、2017年にversion 7の論文が出ています。
- Webサイト:OpenMM
- GitHub:openmm
- ドキュメント:User Guide
Webサイト上でチュートリアルや動画が公開されていたり、OpenMMを利用したソフトウェア・プロジェクトの紹介がされていたりと、ライブラリのエコシステムの維持・発展を意識した運営がされているのがとても魅力的です。
また、初学者にとって嬉しいことにシミュレーション条件をセットアップするためのGUIツールOpenMM Setupも提供されています。

英語 & CUI だと諦める私にとっては最高です!
2-2. OpenMMのインストール
condaがあればOpenMMのインストールは簡単です。(OpenMMユーザーガイド-Installing OpenMM)*1
conda install -c conda-forge openmm
GPUがある場合はインストール時にドライバをオプションで設定できるそうです。(私はMac Book Pro 2017でNvidia GPUもAMD GPUもないです。。。)
インストールがうまくいったかどうかは次のコマンドで確認できます。ターミナルにコピペ。
python -m openmm.testInstallation
こんなのでました。
OpenMM Version: 7.7 Git Revision: 130124a3f9277b054ec40927360a6ad20c8f5fa6 There are 3 Platforms available: 1 Reference - Successfully computed forces 2 CPU - Successfully computed forces 3 OpenCL - Successfully computed forces Median difference in forces between platforms: Reference vs. CPU: 6.3052e-06 Reference vs. OpenCL: 6.7302e-06 CPU vs. OpenCL: 9.02393e-07 All differences are within tolerance.
Successfullyっていってるからたぶん大丈夫。
2-3. OpenMM Setupのインストール
同様にOpenMM Setupもcondaでインストールできます。
conda install -c conda-forge openmm-setup
インストールできたら以下のコマンドでGUIが立ち上がります。
openmm-setup
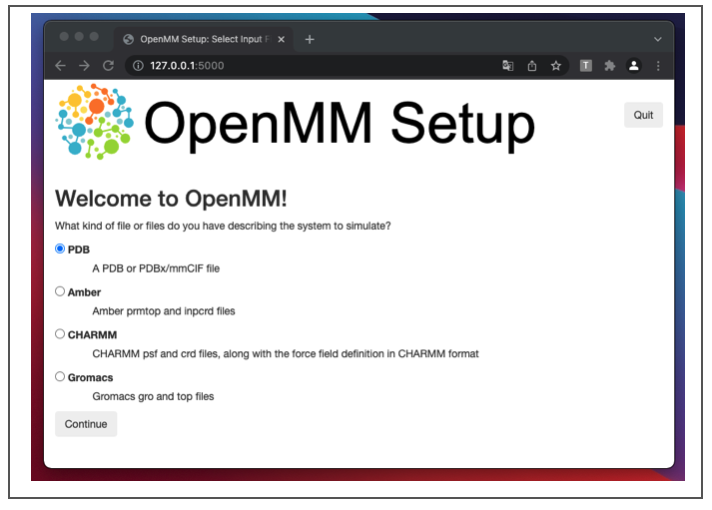
ブラウザで立ち上がりました!ターミナルには以下のような表示が出ました。
* Serving Flask app 'openmmsetup.openmmsetup' (lazy loading) * Environment: production WARNING: This is a development server. Do not use it in a production deployment. Use a production WSGI server instead. * Debug mode: off * Running on http://127.0.0.1:5000/ (Press CTRL+C to quit)
ブラウザが自動で開かなくても「http://127.0.0.1:5000/ 」をコピペしてアクセスすればOKで、終了したければ「CTRL + C」だそうです。Jupyter notebook使う時と一緒ですね。
このまま遊びたいところですが話が逸れるので次回にお預け。
3. 分子力場プロジェクト Open Force Field
3-1. 概要
つづいて分子力場のプロジェクトOpen Force Field(OpenFF)のご紹介です。
分子力場自体はOpenMMのツール群にも用意されていて、OpenMM ForceFields(GitHub)で利用することができます。こちらを利用することで、生体分子をメインにしたAmberやCHARMMといった分子力場や、低分子のためのGAFFやSMIRNOFFといった分子力場を使えるようになります。
なお、OpenMM ForceFieldsもOpenMM本体同様、condaでインストールできます。
conda install -c conda-forge openmmforcefields
で、OpenMM ForceFieldsで提供されている低分子の分子力場SMIRNOFFを作成したのがOpen Force Field イニシアチブです。
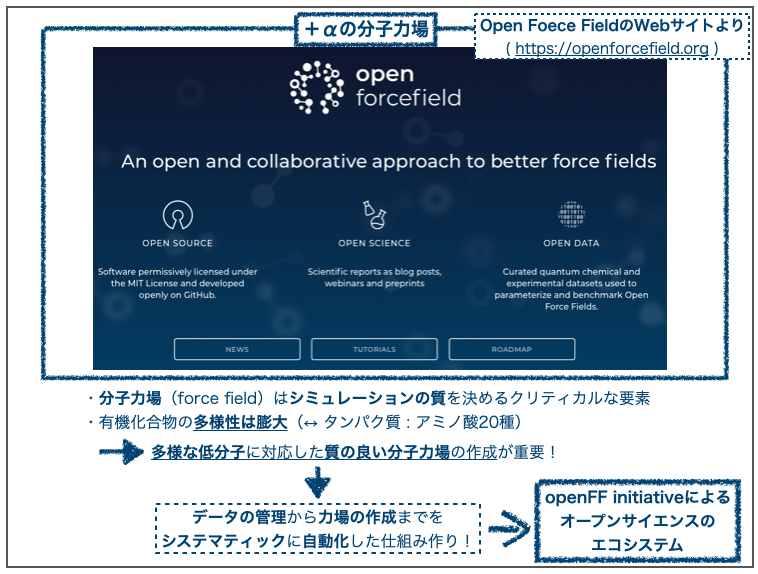
タンパク質の構成要素は凡そ20種に還元できますが、低分子は原子の種類・結合パターンがより多様で複雑です。低分子を対象にシミュレーションを行うには、広大なケミカルスペースに対応した精度の高い分子力場の作成が肝になります。
科学の発展(ex. 新規合成方法の開発)に伴ってケミカルスペースは継続的に発展しつづけるため、対応する分子力場も継続的に改善していく仕組み(エコシステム)作りが求められます。
「それならオープンサイエンスの出番でしょ!」
ってことで(?)、できたイニシアティブがOpen ForceField initiativeです。基になるデータ管理から力場の作成までをオープンかつ再現性よく行うため、システマティックに自動化した仕組みづくりが目指されています。

Open Force Field v.1.0.0(コードネーム Parsely)の論文は下記でプレプリントでもチェックできます。
DEVELOPMENT AND BENCHMARKING OF OPEN FORCE FIELD V1.0.0, THE PARSLEY SMALL MOLECULE FORCE FIELD Qiu, Y., Smith, D., Boothroyd, S., et al., J. Chem. Theory Comput. 2021, 17, 10, 6262–6280
chemRxiv DOI: https://doi.org/10.26434/chemrxiv-2021-l070l-v4 (CC BY 4.0)
Open Force Fieldで作成された力場は「SMIRKS-native Open Force Field(SMIRNOFF)」と名付けられています。SMIRNOFFのパラメータは量子力学(QM)計算で作成したデータセットに対してフィッティングされています。各パラメータの定義は、相互作用のタイプ(ex. 結合の伸縮)とSMIRKSパターンにそれぞれ基づいています。
SMIRKSは化学構造の線形表記の一種(SMILESとSMARTSのハイブリッド?)で、反応前後における分子の変化を一般化して記述できる表記方法のようです*2。論文のFigure 11を見るとイメージしやすいので引用しておきます。
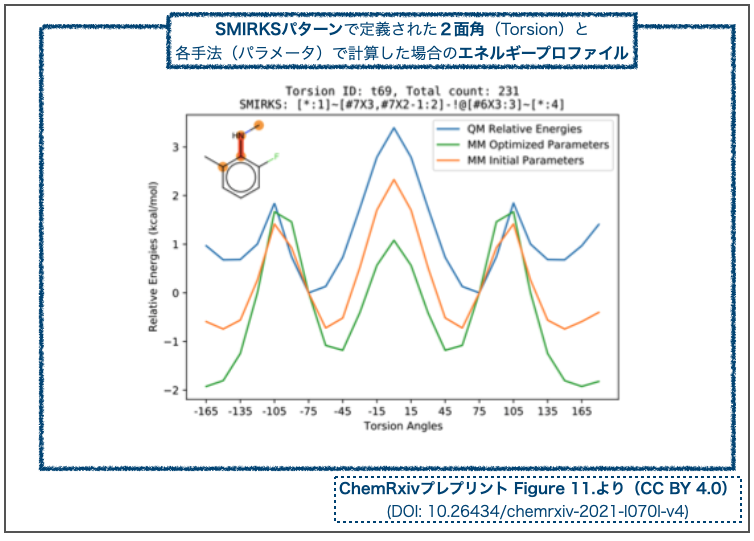
上図はグラフ左上の構造式でハイライトされた4原子の2面角の、角度に依存したエネルギー変化のプロットです。プロット上部の「SMIRKS」部分をみれば、パターンで部分構造を定義しているというのがわかりますね。
力場作成の詳細な方法や精度のベンチマークは論文をご参照ください。
3-2. インストール
Open Force Fieldを利用するにはOpen Force Field ToolkitをインストールすればOKです。condaがあれば簡単です。(openff toolkit - installation)
conda install -c conda-forge openff-toolkit
ドキュメントには使い方の実例がたくさん載っています(Examples using SMIRNOFF with the toolkit)。GitHubのレポジトリにリンクされていて、ノートブックやBinderでのデモも用意されているので要チェックだ!
…私はまだ一つも見れてません。すみません。
4. 準備ツール
つづいて、シミュレーションを行うための準備に関するツールです。モデル構造の修正(PDBFixer)や、ファイルフォーマットの変換(ParmED)といった操作に使えます。
4-1. PDBFixerでPDB構造を修正
タンパク質のシミュレーションを行う際、基になる構造情報をPDBから取得するケースが多いと思います。ですが、PDBに登録されている情報が必ずしもシミュレーションに適しているとはいえないため、目的に合わせた修正を行う必要が生じます。
PDBFixerはシミュレーションに合わせたPDBファイルの修正(fix)を行うためのツールで、OpenMMのエコシステムに組み込まれています。
こんな感じのことができます。

PDBFixerのインストールもcondaでOKです。
conda install -c conda-forge pdbfixer
PDBFixerはコマンドラインアプリケーションとしても使えますが、Python APIが用意されていてそちらの方がpowerfulに使えるそうです。
4-2. ParmEdでフォーマットを変換
計算で遊ぶときの一つのネックは、色々ソフトウェアがあって、それぞれに異なる拡張子のファイルを使っている場合です。MDシミュレーションにもAmberやCHARMMといった複数のソフトがあります。
ParmEdは異なるファイルフォーマット間で変換や読み書きができ、この問題を解決してくれます。生体分子シミュレーションの主要なソフトウェアをカバーしていて、Open Force Field Toolkitの使用例でも構築した系の変換に使われています(Export OpenFF-generated OpenMM System to AMBER and GROMACS files)。

- GitHub : ParmEd
- ドキュメント : ParmEd
- 文献:Shirts, M.R., Klein, C., Swails, J.M. et al. Lessons learned from comparing molecular dynamics engines on the SAMPL5 dataset. J Comput Aided Mol Des 31, 147–161 (2017). DOI: 10.1007/s10822-016-9977-1
ParmEdのインストールもcondaでOKです。
conda install -c conda-forge parmed
本記事で参考にしている資料では、Making it rainのProtein-Ligand Simulationsで、タンパク質-リガンド複合体のシミュレーション系を構築する際に利用されていました。
5. 結果(トラジェクトリ)の解析
OpenMMはMD計算の準備と実行、結果の出力までできますが、得られた結果(トラジェクトリ)の解析には、それに特化したソフトウェアがいくつかあるようです。参考資料の中にはPyTraj、MDTraj、MDAnalysisというツールがでてきました。・・・そもそもトラジェクトリ解析にどのような手法があるのかわかっていないのでツール間の違いがよく分かりませんでした。
5-1. PyTraj
PyTrajはAMBERのAmber Toolsに組み込まれているcpptrajというプログラムをPythonから扱えるようにしたフロントエンドパッケージです。MDでよく見るAMBERプロジェクトと関わりがある点が魅力的です。
- GitHub: pytraj
- ドキュメント:pytraj 2.0.2.dev0
インストールはcondaでできますが、チャンネルがambermdである点に注意です(pytraj - Installation)。
conda install -c ambermd pytraj
ドキュメントにチュートリアルをたくさん用意してくださっているので、こちらをみれば大体どんなことができるのか分かりそうです。

参考資料では、Making it rainのトラジェクトリ解析(RMSD計算、PCA解析 etc.)は基本的にPyTrajで実行されています。
また、他の解析パッケージとの互換も意識されてるようで、ドキュメントにはmdtrajやmdanalysisとのインターフェイスについてのチュートリアルがあります。
5-2. MDTraj
つづいてMDTrajです。こちらもPythonで利用可能なトラジェクトリ解析ツールです。OpenMMの開発も行っているStanford大学のPande Labによるプロジェクトという点で魅力的です*3。
- GitHub : mdtraj
- ドキュメント:MDTraj
- 文献:McGibbon, R.T., Beauchamp, K.A., Harrigan, M.P., et. al., MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories Biophysical Journal 2015(109)1528–1532 DOI: 10.1016/j.bpj.2015.08.015
MDTrajもcondaでインストールできます(MDTraj - Installation)。
conda install -c conda-forge mdtraj
こちらもドキュメントに使用例がコードとともに掲載されています。
参考資料では、TeachOpenCADDのT019 · Molecular dynamics simulationで使われていてます。トラジェクトリの解析ではなく、それぞれ別に用意したタンパク質とリガンドのモデルを一つの複合体としてシミュレーションするためにマージするツールとして使われています。
5-3. MDAnalysis
さいごにMDAnalysisです。これもPythonで利用可能なトラジェクトリ解析ライブラリです。ドキュメントだけでなくブログやビデオチュートリアルがあったりと、開発が活発に続けられている感じなのが素敵です。
- GitHub:MDAnalysis
- ドキュメント:MDAnalysis documentation
- 文献①:R. J. Gowers, M. Linke, J. Barnoud, T. J. E. Reddy, M. N. Melo, S. L. Seyler, D. L. Dotson, J. Domanski, S. Buchoux, I. M. Kenney, and O. Beckstein. MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations. In S. Benthall and S. Rostrup, editors, Proceedings of the 15th Python in Science Conference, pages 102-109, Austin, TX, 2016. SciPy. doi:10.25080/Majora-629e541a-00e
- 文献②:N. Michaud-Agrawal, E. J. Denning, T. B. Woolf, and O. Beckstein. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 32 (2011), 2319--2327. doi:10.1002/jcc.21787
MDAnalysisもcondaでインストールできます(MDAnalysis - Installation)。
conda install -c conda-forge mdanalysis
JupyterLabで使いたい場合は、合わせてnodejsをインストールして設定しておくと良いそうです(解析の際にプログレスバーが表示できたりするらしい?)。
conda install -c conda-forge nodejs jupyter labextension install @jupyter-widgets/jupyterlab-manager
使用例やチュートリアルはMDAnalysis - User Guideに詳しいです。
参考資料では、TeachOpenCADDのT020 · Analyzing molecular dynamics simulationsでトラジェクトリ解析に使われています。RMSDの計算やタンパク質-リガンド間の相互作用(原子間距離、水素結合)の解析が行われています。
6. おわりに
以上、今回はオープンソースソフトウェアを使ってMD計算を試す際に便利そうなツールをひろってみました。といってもMaking it rainとTeachOpenCADDで出てくるソフトを列挙しただけですが、、、
「実際にどう使うか?」までは踏み込めず、「(似たような)ツール間の違い」も分かりませんでしたが、とりあえずドキュメントのありかがわかった!!
複雑な計算科学の分野で、オープンサイエンスにむけた取り組みが進んでいるのはとてもありがたい話ですね。エコシステムとして維持・発展させるための取り組みには科学に限らず参考にすべき点が多い気がします。・・・まず、かわいいロゴが大事!!
オープンマインドにあふれた偉大な先人たちのおかげで、いろいろなドキュメントやチュートリアルが手に入るようになりました。・・・が、残念なことにレベルが低い私はノートブックの最初に列挙されているimport ~~ as ~~が追いきれず、途中で「これなんのライブラリ使ってるんだ??どっから来た?」「自分で遊ぶときには何をインストールして使えばいいんだ???」 となってしまいます。
今回、なんとなくMD計算で利用できそうなソフトの名前がわかったのでヨシ!
他にもおすすめのソフトウェアがあったら教えていただけると嬉しいです。間違いが多そうなのでご指摘いただければ幸いです。
ではでは。