OpenMMをステップバイステップで 〜Part 8 低分子-タンパク複合体シミュレーション〜
OpenMMの使い方を順番にたどる記事のPart 8です。
Part 7まででタンパク質単体のMDシミュレーションについて、「①準備(前処理)、②実行、③結果の解析」の流れがなんとなくわかりました。第1ステージはクリア!…ということにして、第2ステージ「低分子 - タンパク質複合体」のシミュレーションを試したいと思います。
この記事ではTeachOpenCADD T019:Molecular dynamics simulationで行われている「複合体シミュレーション系の構築と計算」の理解を目指します。題材にする構造はPDB ID: 3POZでEGFRタンパクとリガンドの複合体です。
低分子リガンドのための力場としてopen forcefieldを利用し、OpenMMのシミュレーション系を構築します。計算はGoogle Colabで行いました。*1

1. 低分子を含むMDシミュレーションに向けた準備
1-1. タンパク質単独と何が違う?
最初にタンパク質単独と低分子-タンパク質複合体のシミュレーション準備で違う点です。
一番大きな違いは力場です。タンパク質の構造は基本的に(限られた種類の)アミノ酸からなりますが、低分子リガンドはより多様な原子タイプ・結合パターンを含んでいます。なのでより広い領域をカバーした力場パラメータが計算に求められます。
OpenMMにデフォルトで含まれる力場パラメータは生体分子(タンパク質、DNA、RNA ...)や水モデルには対応していますが、低分子には対応していません。そこで別に低分子の力場を作成して取り込んでやる必要があります。
ここではOpenMM Forcefieldsを経由してopen forcefieldの力場を利用します。
1-2. OpenMM Forcefields?
OpenMM Forcefields はOpenMMの追加の力場を提供しているツールです(GitHub)。
サポートしている力場は① AMBER、② CHARMM、③ Open Force Field Initiative force fields です。
低分子についてはAMBERのGeneral AMBER Force Field(GAFF)やOpen Force Fieldのopenff、smirnoffといった力場が使えるようになります。
インストールはcondaでOKです。
conda install -c conda-forge openmmforcefields
1-3. Open Force Field Initiative?
似たような名前が多くて混乱しますが、Open Force Field InitiativeはOpenMMとは別の分子力場に焦点を当てたプロジェクトです。以前こちらのブログ記事でも取り上げました。
継続的な分子力場の改善を目指したイニシアティブで、基になるデータ管理から力場の作成までをオープンかつ再現性よく行うため、自動化した仕組みづくりを目指しているそうです。

現在(2022/05)、以下の3系統の力場が提供されています。

左から右に新しくなります。 SMIRNOFF(スミノフ?)はSMIRKS-native Open Force Fieldの略で、「OpenFF 1.y.x」系統のコードネームがParsley(パセリ)、「OpenFF 2.y.x」系統のコードネームがSage(セージ)です。*2
GitHubへのリンクはそれぞれ以下です。
利用するにはOpen Force Field ToolkitをインストールすればOKです。condaがあれば簡単です。
conda install -c conda-forge openff-toolkit
使える力場パラメータのバージョンは以下で確認できます。
from openff.toolkit.typing.engines.smirnoff import forcefield print(forcefield.get_available_force_fields()) # ['smirnoff99Frosst-1.0.2.offxml', 'smirnoff99Frosst-1.0.0.offxml', 'smirnoff99Frosst-1.1.0.offxml', 'smirnoff99Frosst-1.0.4.offxml', 'smirnoff99Frosst-1.0.8.offxml', 'smirnoff99Frosst-1.0.6.offxml', 'smirnoff99Frosst-1.0.3.offxml', 'smirnoff99Frosst-1.0.1.offxml', 'smirnoff99Frosst-1.0.5.offxml', 'smirnoff99Frosst-1.0.9.offxml', 'smirnoff99Frosst-1.0.7.offxml', 'openff-1.0.1.offxml', 'openff-1.1.1.offxml', 'openff-1.0.0-RC1.offxml', 'openff-1.2.0.offxml', 'openff-1.3.0.offxml', 'openff_unconstrained-2.0.0-rc.1.offxml', 'openff_unconstrained-1.3.1.offxml', 'openff_unconstrained-1.2.1.offxml', 'openff-2.0.0-rc.2.offxml', 'openff_unconstrained-1.0.0-RC2.offxml', 'openff_unconstrained-1.1.0.offxml', 'openff_unconstrained-1.0.0.offxml', 'openff-2.0.0.offxml', 'openff_unconstrained-2.0.0.offxml', 'openff_unconstrained-2.0.0-rc.2.offxml', 'openff-1.1.0.offxml', 'openff-1.0.0.offxml', 'openff-1.0.0-RC2.offxml', 'openff-1.3.1.offxml', 'openff-1.2.1.offxml', 'openff-1.3.1-alpha.1.offxml', 'openff_unconstrained-1.0.0-RC1.offxml', 'openff_unconstrained-1.2.0.offxml', 'openff_unconstrained-1.3.0.offxml', 'openff-2.0.0-rc.1.offxml', 'openff_unconstrained-1.0.1.offxml', 'openff_unconstrained-1.1.1.offxml', 'openff_unconstrained-1.3.1-alpha.1.offxml']
ファイル形式はoffxmlのようです。OpenFFのxmlという感じでしょうか?
OpenFF toolkitがあるとRDKitのMolオブジェクトをOpenFFで使えるように変えたりすることもできます。
1-4. MDTrajのインストール
準備の最後にMDTrajをインストールしておきます。

こちらはTrajとついている通りトラジェクトリ解析用のツールです。前回の記事でご紹介した埼玉大学 松永先生のコロキウム(生体分子系の分子動力学シミュレーションデータの解析入門)でも、ファイルの拡張子変換機能(mdconvert)が使いやすいと紹介されていました。
今回の記事では、別々に前処理したタンパク質と低分子リガンドを一つの複合体にまとめる(マージする)ツールとして利用します。
インストールはcondaでOKです。
conda install -c conda-forge mdtraj
これでツールの準備はおしまいです。
(省略しましたがRDKitも使います。*3)
2. 計算の対象構造について
具体的なシミュレーションの準備を行う前に、計算の対象の構造を確認しておきます。
今回計算の対象とする低分子-タンパク質複合体の構造はTeachOpenCADD T019:Molecular dynamics simulationを踏襲してPDB ID: 3POZです。
EGFRタンパクのキナーゼドメインとリガンド TAK-285のX線共結晶構造です。*4

文献はこちら
Aertgeerts K, Skene R, Yano J, et.al., Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J Biol Chem. 2011(286)18756-65. doi:10.1074/jbc.M110.20619351133-2/fulltext)
複合体構造の中でのTAK-285のLigand IDは03Pです。あとでPDBファイルから構造を取り出す際に使います。*5
3. タンパク質-低分子複合体の系をつくろう!
それでは具体的な処理を進めていきましょう!
この記事では操作の理解を重視するため、TeachOpenCADDでは関数にまとめて定義してある部分を解体して眺めていきます。
3-1. 処理全体の流れ
最初に処理の流れを確認します。以前の記事(part 5 シミュレーションプログラムの中身を理解しよう)で取り上げましたが、OpenMMのシミュレーションプログラムはざっくり以下のようなクラス構成です。
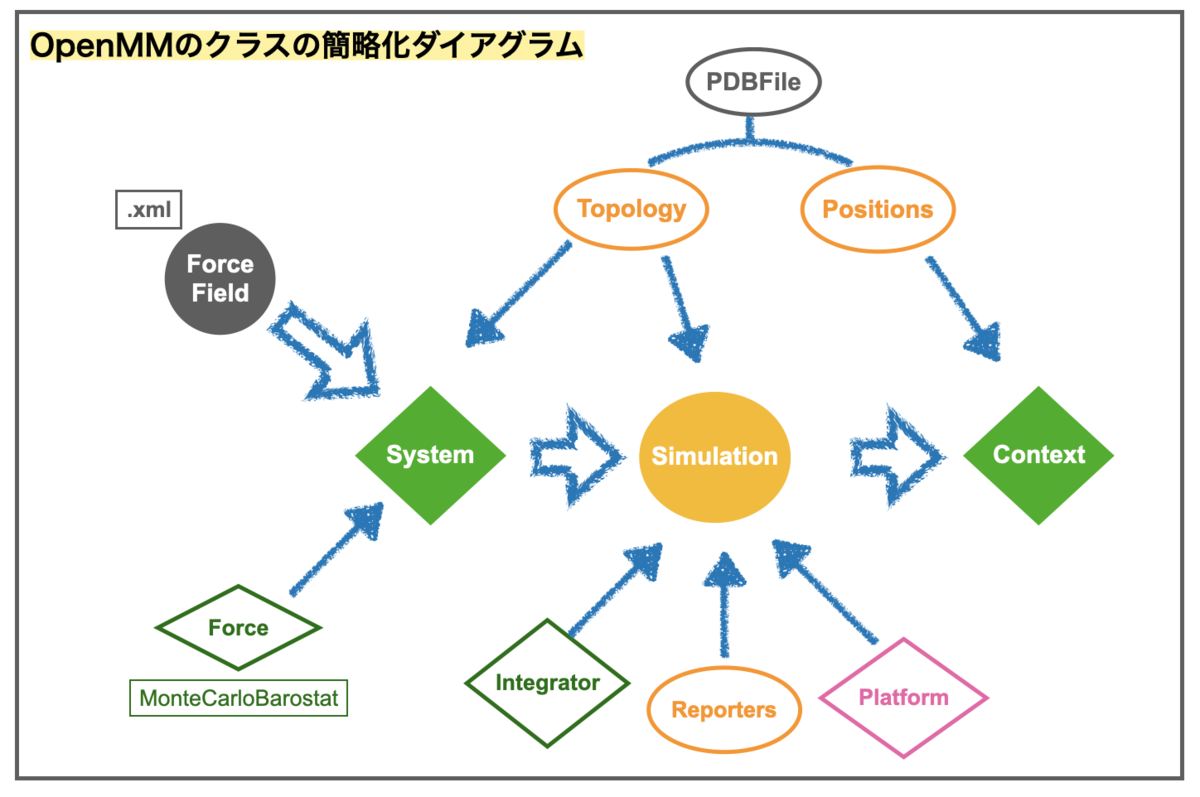
今回、低分子リガンドを加えるにあたって処理を増やすのは左半分です。
追加する処理を加えてみました(水色)。

タンパク質-リガンド複合体のPDBファイルから「①リガンド部分を抜き出し」て、「② Open Forcefieldで力場を設定」し、「③ タンパクとマージ」して複合体に戻します。
より具体的に、処理と使うツールは以下のようになります。
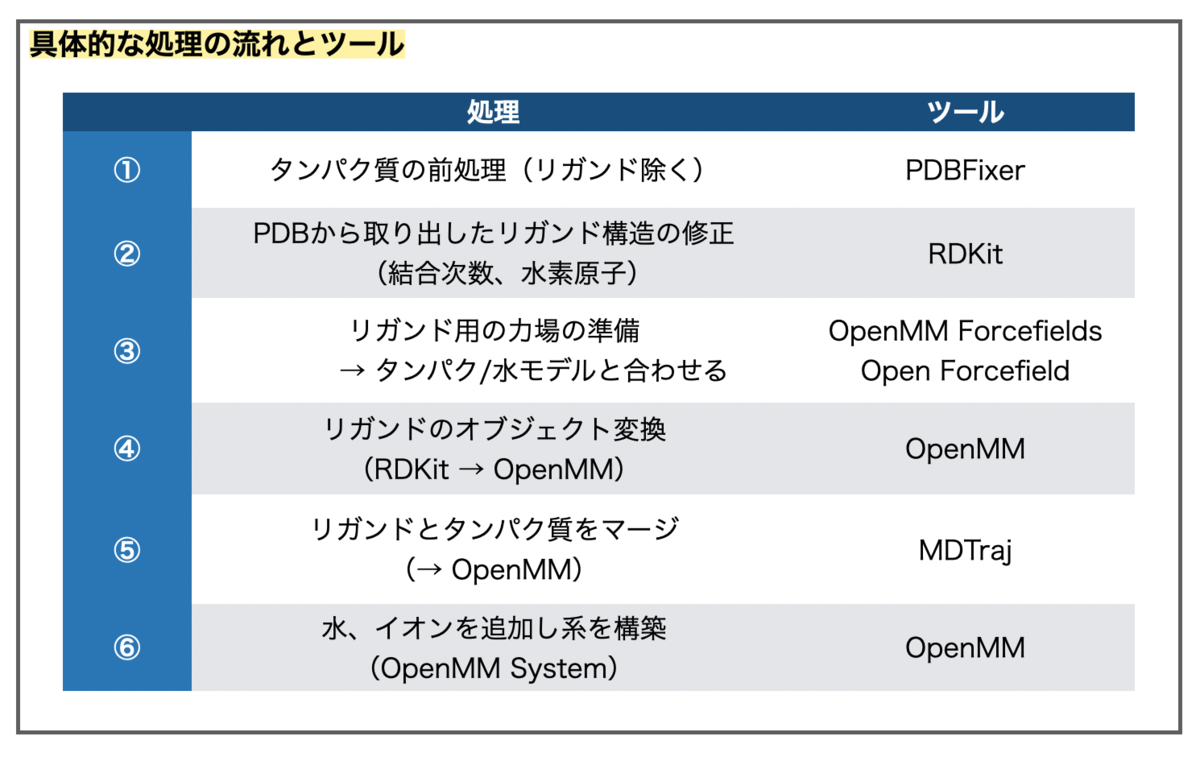
PDBファイルには低分子リガンドの結合次数や水素原子の情報が含まれていないので、これらを補完するためにRDKitを使います。Open Forcefieldを使って低分子の力場を補充し、タンパク-低分子の複合体の系を構築し、最後にシミュレーションの箱(水、イオン追加)を用意して設定完了です。
3-2. Step 1:タンパク質の前処理
Step ①はタンパク質部分の前処理です。ここで行う作業は以前の記事(「Part 2 PDBFixerでタンパク質の前処理」)で取り上げた内容とほぼ同じです。
PDBFixerを使って以下のような作業をします。

水やイオンの追加はここではまだ行わないことにご注意ください*6。
こんな感じのコードです。*7
# 処理したいPDBファイルの指定 pdb_file = 'Data/3poz.pdb' # 必要なライブラリのインポート import pdbfixer # PDBFixerでPDBファイルを読み込む fixer = pdbfixer.PDBFixer(pdb_file) # 不要な構造の削除 fixer.removeHeterogens() # 欠けている残基のチェック(欠損原子の確認のためにも必要) fixer.findMissingResidues() # タンパク質末端の欠けている残基を取り除く処理 chains = list(fixer.topology.chains()) keys = fixer.missingResidues.keys() for key in list(keys): chain = chains[key[0]] if key[1] == 0 or key[1] == len(list(chain.residues())): del fixer.missingResidues[key] # 非標準な残基が含まれているか確認、あれば標準的なものに置き換える fixer.findNonstandardResidues() fixer.replaceNonstandardResidues() # 欠けている原子の確認、あれば追加する fixer.findMissingAtoms() fixer.addMissingAtoms() # 水素原子の付与(pHを設定する) ph = 7.0 fixer.addMissingHydrogens(ph) print(type(fixer)) # <class 'pdbfixer.pdbfixer.PDBFixer'>
得られたPDBFixerのオブジェクトには処理後のタンパク質の原子の座標(positions)とトポロジー(topology)が含まれています。
3-3. Step 2:リガンド構造の修正
続いてリガンド構造の準備です。
PDBから構造や座標を取り出しますが、結合次数や水素原子の情報がPDBファイルには含まれていません。そこでこれらの情報をRDKitを利用して追加します。
まず、RDKitでPDBファイルを読み込んで残基ごとにスプリット(SplitMolByPDBResidues)し、リガンド(ID:03P)を抜き出します。
from rdkit import Chem from rdkit.Chem import AllChem # PDBファイルを読み込んで残基ごとにスプリット rdkit_pdb = Chem.MolFromPDBFile('Data/3poz.pdb') rdkit_pdb_split = Chem.SplitMolByPDBResidues(rdkit_pdb) # 残基 03Pの抜きだし pdb_tak285 = rdkit_pdb_split["03P"] Chem.Draw.MolToImage(pdb_tak285)

単結合だけだと寂しい感じ・・・。
SMILES情報からTAK-285のリファレンス構造を作成し、リファレンスをもとに結合次数を追加しましょう(AssignBondOrdersFromTemplate])。
SMILESはRCSB PDBのリガンドページ(03P)から持ってきました。
# 中途半端に残ってる水素原子を消しておく pdb_tak285_noH = Chem.RemoveHs(pdb_tak285) # SMILESからリファレンス構造を作成 smi_tak285 = "CC(C)(CC(=O)NCCn1ccc2c1c(ncn2)Nc3ccc(c(c3)Cl)Oc4cccc(c4)C(F)(F)F)O" ref_tak285 = Chem.MolFromSmiles(smi_tak285) # リファレンスをもとに結合次数をアサイン prep_tak285 = AllChem.AssignBondOrdersFromTemplate(ref_tak285, pdb_tak285) prep_tak285.AddConformer(pdb_tak285.GetConformer(0)) # 水素原子の付加 prep_tak285 = Chem.AddHs(prep_tak285, addCoords=True) Chem.Draw.MolToImage(prep_tak285)

3-4. Step 3:リガンド用の力場の準備
リガンド構造が用意できたので力場を準備します。
最初にOpenMMでタンパク質(amber14-all.xml)と水モデル(amber14/tip3pfb.xml)の力場を設定しておきます。
from openmm import * from openmm.app import * FF = ForceField('amber14-all.xml', 'amber14/tip3pfb.xml')
このForceFieldに低分子の力場を追加します。
まず、リガンドを「RDKitのMolオブジェクト」から「OpenFFのMoleculeオブジェクト」に変更します。openff toolkitのMolecule.from_rdkitを使えばOKです。*9
from openff.toolkit.topology import Molecule off_tak285 = Molecule.from_rdkit(prep_tak285, allow_undefined_stereo=True, hydrogens_are_explicit=True) print(type(off_tak285)) # <class 'openff.toolkit.topology.molecule.Molecule'>
OpenMMのForceFieldに低分子の力場を追加するには、低分子の構造から① Templateを作ってそれを②ForceFieldにregisterします。
今回はOpen Forcefieldに用意されているGAFFを力場に使います。この場合は「① GAFFTemplateGenerator」と「② registerTemplateGenerator」を使うことになります。
from openmmforcefields.generators import GAFFTemplateGenerator gaff_temp = GAFFTemplateGenerator(off_tak285) FF.registerTemplateGenerator(gaff_temp.generator)
なお、GAFFはAMBER力場の仲間で、低分子医薬品デザインのために作られたものだそうです。タンパク質側をAMBER力場で設定しているときに互換性の良い低分子力場となる感じでしょうか?
今回使用したGAFFのバージョンや、Open forcefieldにインストールされているGAFFのバージョン一覧は以下のように確認できます。
print("version info : ", gaff_temp.gaff_version) print("installed versions : ", GAFFTemplateGenerator.INSTALLED_FORCEFIELDS) # version info : 2.11 # installed versions : ['gaff-1.4', 'gaff-1.8', 'gaff-1.81', 'gaff-2.1', 'gaff-2.11']
使われたバージョン 2.11のようです。
これで力場の準備が終わりました。
3-5. Step 4:リガンドのオブジェクト変換
つづいてリガンドのオブジェクトを変換します。タンパク質とマージするための準備です。
力場を作る際にRDKitからOpenFFのMoleculeオブジェクトにしましたが、これをさらにOpenMMのオブジェクトにします。
まず、OpenFF Moleculeオブジェクトに、分子および原子の名前を追加します。原子の名前は「元素記号 + 数字」形式です。
# 分子の名前("LIG")を追加 off_tak285.name = "LIG" # 原子の名前を追加 # 辞書を用意しておいて、元素記号+数字形式にする element_counter_dict = {} for off_atom, rdkit_atom in zip(off_tak285.atoms, prep_tak285.GetAtoms()): element = rdkit_atom.GetSymbol() if element in element_counter_dict.keys(): element_counter_dict[element] += 1 else: element_counter_dict[element] = 1 off_atom.name = element + str(element_counter_dict[element])
RDKitのGetSymbolで原子の元素記号を一つずつ取り出して辞書に格納しています。同じ元素記号があったら数字を+1するので、炭素なら「C1、C2・・・」と順番に原子に名前がつけられていきます。
名前をつけたらOpenFFからOpenMM(topology、positions)に変換します。ひとつ注意点として、OpenMMの単位はnm基準なので、PDBファイルのオングストロームから変更する必要があります。
# OpenMMの単位系導入 from openmm import unit # トポロジー(topology)の取り出しと変換 off_top = off_tak285.to_topology() omm_top = off_top.to_openmm() # 座標(positions)の取り出し omm_pos = off_tak285.conformers[0] # 単位をÅからnmへ for atom in omm_pos: coords = atom / atom.unit atom = (coords / 10.0) * unit.nanometers # TopologyとPositionsをまとめてOpenMMのオブジェクトに変換 omm_tak285 = app.Modeller(omm_top, omm_pos)
最後にtopologyとpositionsをまとめてOpenMMのModellerオブジェクトにしています。
これでリガンド側での準備はもろもろ完了です。タンパク質と合体しましょう!
3-6. Step 5: リガンドとタンパク質のマージ
ここまででタンパク質のPDBFixerオブジェクトとリガンドのModellerオブジェクトが用意できました。これらをまとめて一つにします。
topologyとpositionsを別々に処理します。
まずtopologyですが、MDTrajを利用します。タンパク質とリガンドをそれぞれMDTrajのオブジェクトに変換(Topology.from_openmm)したのちjoinで結合し、OpenMMオブジェクトに戻します(to_openmm)。
import mdtraj as md # タンパク質とリガンドのトポロジーをそれぞれMdTrajに変換 md_protein_top = md.Topology.from_openmm(fixer.topology) md_ligand_top = md.Topology.from_openmm(omm_tak285.topology) # joinでひとつのトポロジーにまとめる md_complex_top = md_protein_top.join(md_ligand_top) # OpenMMオブジェクトにする omm_complex_top = md_complex_top.to_openmm()
ついで、positionsです。
各原子の位置の値と単位(nm)を含むタプルの配列を作成します。OpenMMのQunatityクラスとなります。
import numpy as np # タンパクとリガンドを合わせた総数 total_atoms = len(fixer.positions) + len(omm_tak285.positions) # positionsを格納するQuantity配列の準備 complex_positions = unit.Quantity(np.zeros([total_atoms, 3]), unit=unit.nanometers) # タンパク質のpositionsを追加 complex_positions[: len(fixer.positions)] = fixer.positions # 後に続けてリガンドのpositionsを追加 complex_positions[len(fixer.positions) :] = omm_tak285.positions
topologyとpositionsがそれぞれ処理できたので、これらをまとめたOpenMMのオブジェクトにしましょう。
comp_model = app.Modeller(omm_complex_top, complex_positions)
できました!
3-7. Step 6:水、イオンを追加し系を構築
前処理の最後として系に水とイオンを追加します。
水のpaddingの厚みは「1.0 nm」、イオンは「NaCl 0.15 M」とします。水モデルはTIP3Pで、forcefiled(amber14/tip3pfb.xml)で指定されています。
comp_model.addSolvent(FF, padding=1.0 * unit.nanometers, ionicStrength=0.15 * unit.molar)
処理後の状態をPDBファイルで保存しておきましょう。
# 処理後の状態(トポロジー、原子の位置)をPDBファイルで出力 top = comp_model.getTopology() pos = comp_model.getPositions() app.PDBFile.writeFile(top, pos, open('Data/gaff_complex_processed.pdb', 'w'))
こんな構造になりました。

4. 計算条件の設定と実行
4-1. 計算条件の設定
計算系の準備が完了したので、シミュレーションを実行するための設定を行いましょう。
タンパク質単独でシミュレーションした過去記事(「Part 3」)と同様の設定にしたいと思います。
こんな感じです。

コードにするとこんな感じです。
from openmm import * from openmm.app import * from openmm.unit import * # シミュレーションの準備 ## ForceFiledからSystemを構築する system = FF.createSystem(comp_model.topology, nonbondedMethod=PME, nonbondedCutoff=1.0*nanometers, constraints = HBonds, rigidWater=True, ewaldErrorTolerance=0.0005) ## 圧力制御の機能をaddForceする system.addForce(MonteCarloBarostat(1.0*atmospheres, 300*kelvin, 25)) ## 積分計算の設定 integrator = LangevinMiddleIntegrator(300*kelvin, 1.0/picosecond, 0.002*picoseconds) integrator.setConstraintTolerance(0.000001) ## 計算環境をPlatformで設定 platform = Platform.getPlatformByName('CUDA') platformProperties = {'Precision': 'single'} ## simulationの構築 simulation = Simulation(comp_model.topology, system, integrator, platform, platformProperties) simulation.context.setPositions(comp_model.positions) # XML結果出力ための設定 with open("system.xml", mode="w") as file: file.write(XmlSerializer.serialize(system)) with open("integrator.xml", mode="w") as file: file.write(XmlSerializer.serialize(integrator)) # エネルギー最小化の実行 print('Performing energy minimization...') simulation.minimizeEnergy() # 平衡化のステップ print('Equilibrating...') simulation.context.setVelocitiesToTemperature(300*kelvin) simulation.step(50000) # シミュレーション本番の実行の前にreporterオブジェクトを追加する print('Simulating...') dcdReporter = DCDReporter('trajectory.dcd', 10000) dataReporter = StateDataReporter('log.txt', 1000, totalSteps=500000, step=True, speed=True, progress=True, potentialEnergy=True, temperature=True, separator='\t') checkpointReporter = CheckpointReporter('checkpoint.chk', 10000) simulation.reporters.append(dcdReporter) simulation.reporters.append(dataReporter) simulation.reporters.append(checkpointReporter) simulation.currentStep = 0 # シミュレーション本番の実行 simulation.step(500000) # シミュレーション最後の状態をXMLで書き出す simulation.saveState("final_state.xml") # 最後の状態をPDBx/mmcifで出力する設定 state = simulation.context.getState(getPositions=True, enforcePeriodicBox=system.usesPeriodicBoundaryConditions()) with open("final_state.pdbx", mode="w") as file: PDBxFile.writeFile(simulation.topology, state.getPositions(), file)
項目が多いので長いですが、前回同様にメインは「①系の構築 → ② エネルギー最小化 → ③ 平衡化のステップ → ④ トラジェクトリのプロダクションステップ」を行っているだけで、間に結果出力(Reporter)の設定が入っています。
それぞれの操作の意味については以前の記事(「Part 5:シミュレーションプログラムの中身を理解しよう」)をご参照いただければと思います。
4-2. TeachOpenCADDの設定との違い
以上の計算条件の設定は基本的にはTeachOpenCADD T19と同じですが以下の点が違います。
- ① アンサンプル:TeachOpenCADDは圧力制御を加えていない
- ② 非結合相互作用の拘束条件(
constraints、rigidWater) - ③ シミュレーション時間 (平衡化の有無、プロダクションの時間)
- ④ 出力ファイル形式の設定
また、TeachOpenCADDでは変数としてforcefieldやmodellerなどを使っています。これらは同じ名前のメソッドがOpenMMに含まれている場合があり、最初のimportで読み込むmoduleを変えると誤認識される恐れがあります。重複のない変数名に変えた方が無難だともいます。
4-3. GoogleColabで実行する際の注意点
以上の計算条件の設定を実行すればMDシミュレーションが行えるはずですが、1nsの計算をするのは大変です。前回(「Part 4:ColabでOpenMMシミュレーションを実行」)と同様に、今回も計算はGoogle Colabで実行しました。
ひとつ注意点として、前回は「①minicondaをインストール」し、そこに「② condaで色々なソフトウェアをインストール」する手順を取りました。
ですが、今回この方法ではRDKitがうまく動きませんでした*10。
回避策としてTeachOpenCADDを参考にcondacolabを利用しました。
こんな感じ。
!pip install condacolab import condacolab condacolab.install() !conda install -q -y -c conda-forge mdtraj openmm openmmforcefields openff-toolkit pdbfixer rdkit
condacolab.install()を実行すると一度「kernel restart」しますが、気にせず次の「!conda install ~~~」に進んでOKです。
より良い方法をご存知の方教えていただけると嬉しいです。
4-4. シミュレーション動画
結果はこんな動画になりました!
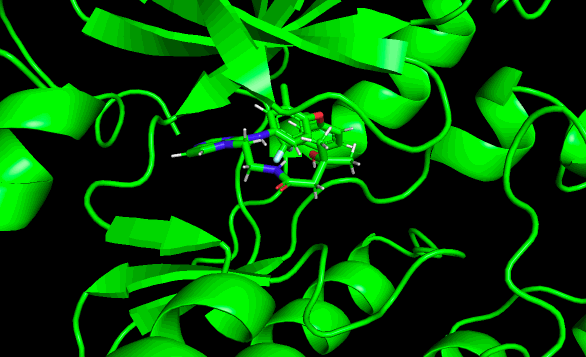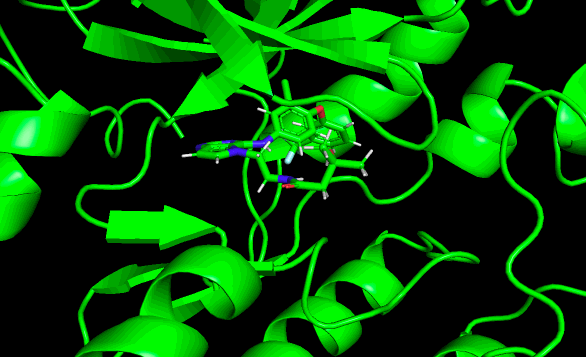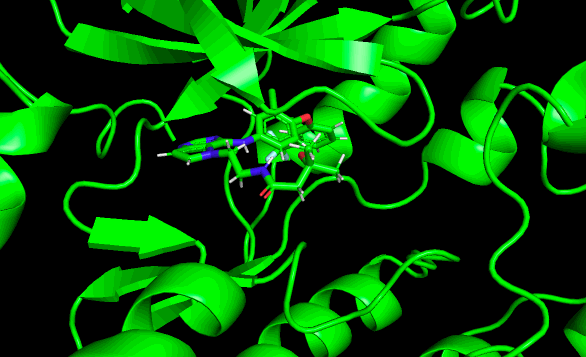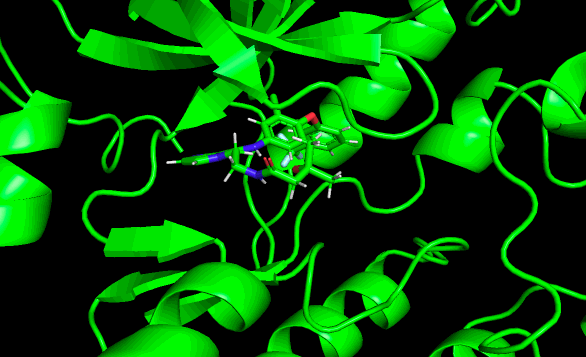
めっちゃ動くやん!
解析は次回!
5. おわりに
以上、今回はMDシミュレーションお勉強の第2ステージということで「低分子 - タンパク質複合体」計算系の設定と実行を行ってみました。
…といってもTeachOpenCADD T19の内容をひとつひとつ確かめただけですが、、、余計にわかりにくくなっていたらごめんなさい。
タンパク質単独のシミュレーションと違って、複合体では低分子リガンドの構造情報の取り扱いや専用の力場の設定が必要になりました。そのため複数のツールを組み合わせて行ったり来たりしなければならず難しかったです。
また、低分子力場を扱うためにOpen Forcefieldを使いました。本当はGAFFではなくSMIRNOFFやOpenFFを使ってみたかったのですが、私の能力では無理でした。
以上、今回も色々と間違いが多いと思います。ご指摘いただければ幸いです。
ではでは!
6. おまけ
低分子の力場としてGAFFではなくOpenFFの利用を試みるとこうなりました。
# 力場の設定・追加 from openmmforcefields.generators import SMIRNOFFTemplateGenerator smirnoff_temp = SMIRNOFFTemplateGenerator(off_tak285) forcefield.registerTemplateGenerator(smirnoff_temp.generator) print("forcefield version : ", smirnoff_temp.forcefield) # forcefield version : openff-2.0.0
最新のOpenFF 2.00(Sage)が使われました。
これを使って進めるとOpenMMで系に「水、イオンを追加(addSolvent)」する際に以下のような出力が出ました。
# 〜〜〜略〜〜〜 <Improper class1="[H][O][C]([C]([H])([H])[H])([C]([H])([H])[H])[C]([H])([H])[C](=[O])[N]([H])[C]([H])([H])[C]([H])([H])[N]1[C]([H])=[C]([H])[c]2[n][c]([H])[n][c]([N]([H])[c]3[c]([H])[c]([H])[c]([O][c]4[c]([H])[c]([H])[c]([H])[c]([C]([F])([F])[F])[c]4[H])[c]([Cl])[c]3[H])[c]21$C22#29" class2="[H][O][C]([C]([H])([H])[H])([C]([H])([H])[H])[C]([H])([H])[C](=[O])[N]([H])[C]([H])([H])[C]([H])([H])[N]1[C]([H])=[C]([H])[c]2[n][c]([H])[n][c]([N]([H])[c]3[c]([H])[c]([H])[c]([O][c]4[c]([H])[c]([H])[c]([H])[c]([C]([F])([F])[F])[c]4[H])[c]([Cl])[c]3[H])[c]21$N5#32" class3="[H][O][C]([C]([H])([H])[H])([C]([H])([H])[H])[C]([H])([H])[C](=[O])[N]([H])[C]([H])([H])[C]([H])([H])[N]1[C]([H])=[C]([H])[c]2[n][c]([H])[n][c]([N]([H])[c]3[c]([H])[c]([H])[c]([O][c]4[c]([H])[c]([H])[c]([H])[c]([C]([F])([F])[F])[c]4[H])[c]([Cl])[c]3[H])[c]21$C20#27" class4="[H][O][C]([C]([H])([H])[H])([C]([H])([H])[H])[C]([H])([H])[C](=[O])[N]([H])[C]([H])([H])[C]([H])([H])[N]1[C]([H])=[C]([H])[c]2[n][c]([H])[n][c]([N]([H])[c]3[c]([H])[c]([H])[c]([O][c]4[c]([H])[c]([H])[c]([H])[c]([C]([F])([F])[F])[c]4[H])[c]([Cl])[c]3[H])[c]21$C23#30" periodicity1="2" phase1="3.141592653589793" k1="1.5341333333333336"/> # 〜〜〜略〜〜〜 <Element Improper at 0x13f32bbc0> # 〜〜〜略〜〜〜
エラーではないようですが、improperとなっているのが気になったので今回は採用しませんでした。
推測に過ぎませんが、OpenFFのSMIRNOFFはSMIRKSベースで2面角の力場を設定しているようなので、適切なクエリに該当しない場合improper判定されるのかもしれません。ご存知の方がいらっしゃったらご教示いただければ幸いです。
*1:ロゴは書くソフトウェアのWebサイトより。イラストはいらすとやさんより。
*2:セージで検索すると薬用サルビアが出てきました。ウォッカからハーブ系統に路線変更したんでしょうか?
*3:このブログを読んでくださっている方は既にインストール済みと思います。
*4:3DビューはRCSB PDB提供のNGL Viewer (AS Rose et al. (2018) NGL viewer: web-based molecular graphics for large complexes. Bioinformatics doi:10.1093/bioinformatics/bty419)
*5:PDBで「Ligand ID:03P」をで検索すると他に3RCD、3W2Oがヒットします。見比べても面白かもしれません。
*6:リガンドを入れる前に水を入れると後でぶつかるから?でしょうか??
*7:詳しい中身は過去記事をご参照いただければと思います。
*8:TeachOpenCADDのコードに従って、結合次数を調整した後、再度AddConformerで元の3D座標を追加していますが、これが必要かはよくわかりません。この操作をしなくてもGetConformer().GetPositions()で確認すると座標は変わらなかったです。
*9:TeachOpenCADDでは引数にhydrogens_are_explicit=Trueが設定されていませんが、デフォルトではFalseで自動的にCham.AddHsを実行することになります。あらかじめ水素原子を付加しているのでTrueにしておきました。
*10:インストール後にimport Chemしようとしたら「必要なGLIBCXX(?)が見つからない」といったエラーでimport出来ませんでした