OpenMMをステップバイステップで 〜Part 7 MDAnalysisでトラジェクトリ解析(②)〜
オープンソースのプログラムでMD計算をお勉強する記事のPart 7です。
今回は前回(part 6)に引き続き、MDAnalysisを使ったシミュレーション結果のトラジェクトリ解析(パート②)を行います。また、解析ツールとしてPyTraj も利用します。
使用するデータはEGFRタンパク質(PDB ID: 3POZ)についてOpenMMで1ns計算したものです。
1. この記事で試す解析
前回の記事では①RMSD、②RMSF、③回転半径の計算を行いました。後半戦では④主成分分析と⑤相互相関解析を行います。

ざっくりと「タンパク質のどのあたりが一緒にうごいているか?」、おおまかな動きの特徴をつかめる解析のようです。
2. 主成分分析(PCA)
2-1. MD計算の主成分分析って?
まずは主成分分析です。統計っぽい話でよくみますがMD計算においてはどのように使われるのでしょうか?
前回の記事でも取り上げさせていただいた埼玉大学 松永 康佑先生のコロキウム動画わかりやすく解説してくださっています(動画の33分あたりからです)。*1
こういう感じのお話?
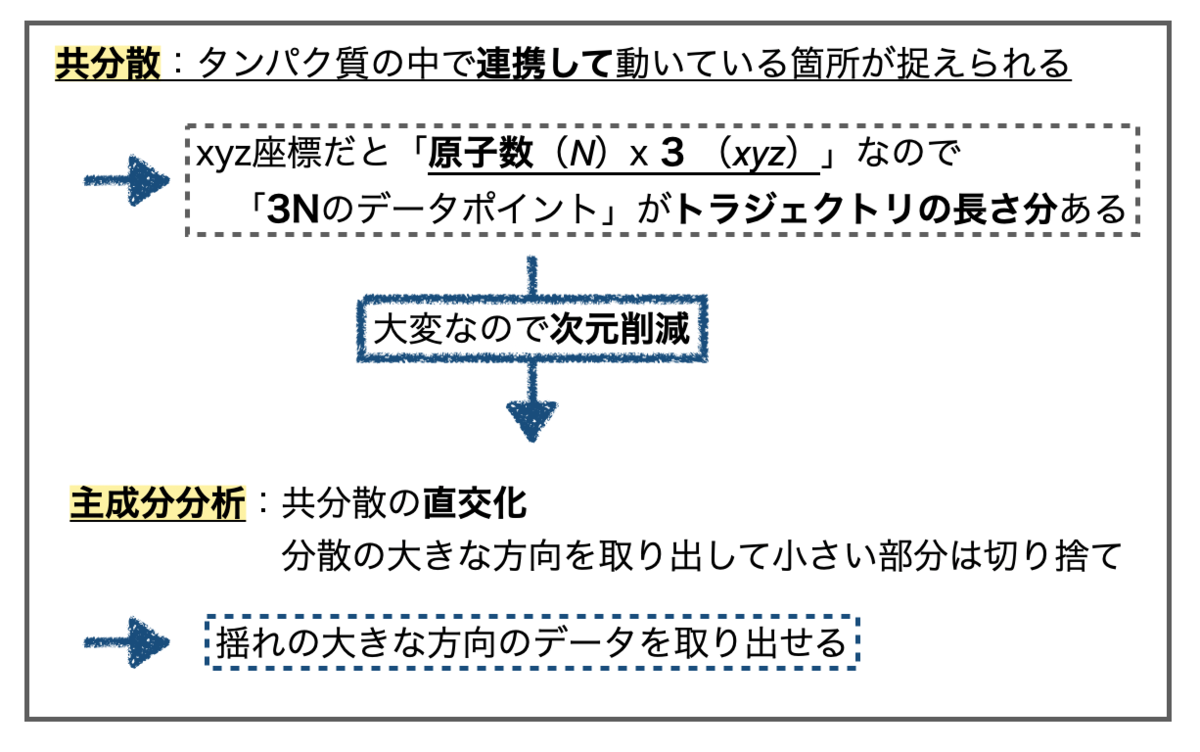
そのままではごちゃごちゃ動いていてよくわからないのが、主成分分析することでノイズを減らして全体としての動きの傾向をつかみやすくなるようです。
上記動画の35分過ぎからは粗視化モデルをつかった主成分分析例が紹介されています。非常にわかりやすいのでおすすめです。
2-2. MDAnalysisで解析の準備(おさらい)
それではMDAnalysisを使った解析を試してみましょう!
前回のおさらいになりますがMDAnalysisはUniverseオブジェクトを中心にデータ解析を行います。topologyとtrajectoryを読み込んで構築します。
import MDAnalysis as mda # topologyとtrajectoryからUniverseオブジェクトを構築 md_universe = mda.Universe("simulation_data/3poz-processed.pdb", "simulation_data/trajectory.dcd") print(type(md_universe))
また、解析に先立ってトラジェクトリのタンパク質のアラインメントをとっておきます(念のため・・)。
from MDAnalysis.analysis import align # トラジェクトリの位置を最初のフレームにリセットする md_universe.trajectory[0] # align.AlignTrajでアラインメントを取る alignment = align.AlignTraj( mobile = md_universe, reference = md_universe, select="protein", in_memory=True) alignment.run()
準備完了!
2-3. MDAnalysisで主成分分析
2-3-1. PCA計算の流れ
主成分分析のやり方については「MDAnalysis User Guide:Principal component analysis of a trajectory」に詳しいチュートリアルがあります。
MDAnalysis.analysis.pca.PCA()を使えば求めることができ、以下のような流れで計算するようです。

それではチュートリアルに従って順番に確認してみましょう。
2-3-2. PCAの実行と累積寄与率の確認
MDAnalysisでPCAを行うにはAnalysisモジュールのMDanalysis.analysis.pca.PCA()を使います。引数にUniverseオブジェクトと目的のAtomGroupの選択基準を渡します。
Cα原子を選択してPCAを行うとこんな感じ。
import MDAnalysis.analysis.pca as pca # Cα原子のPCA Ca_pca = pca.PCA(md_universe, select ='name CA') Ca_pca.run()
PCAの結果の主成分はp_componentsに格納されていて、「原子数 x 3」の配列が成分数分入った配列です(デフォルトで実行した場合は成分数も「原子数 x 3」です。)。
# 主成分を含む配列のサイズ components = Ca_pca.results.p_components print(components.shape) # (951, 951)
シミュレーションの対象としているタンパク質の構造は317残基(Cα原子:317個)なのでちょうど3倍の951になりました。
分散(variance)および累積寄与率(cumulated_variance)を確認してみます。どちらもnumpy.ndarrayなのでインデックスを指定して取り出せます。
# 寄与率 print("1st varinace : ", Ca_pca.results.variance[0]) print("2nd varinace : ", Ca_pca.results.variance[1]) # 累積寄与率 print("1st cumulative varinace : ", Ca_pca.results.cumulated_variance[0]) print("2nd cumulative varinace : ", Ca_pca.results.cumulated_variance[1]) # 1st varinace : 63.373490214896336 # 2nd varinace : 14.112964500184088 # 1st cumulative varinace : 0.39144314860346985 # 2nd cumulative varinace : 0.4786156120633174
第1主成分と第2主成分を合わせた累積寄与率は48%でした。
2-3-3. 累積寄与率のプロット
累積寄与率が95% に達するには何番目まで考慮すれば良いでしょうか?
cumulated_varianceはndarrayなのでnp.where()で95より大きくなる要素のインデックスを取得し、その一番最初を取り出します。
import numpy as np import matplotlib.pyplot as plt # 累積寄与率が95%を超える成分 pc_95 = np.where(Ca_pca.results.cumulated_variance > 0.95)[0][0] print(">95% component number : ", pc_95) # 累積寄与率のプロット plt.plot(Ca_pca.results.cumulated_variance[:pc_95]) plt.xlabel("Principal component") plt.ylabel("Cumulative variance") plt.show() # >95% component number : 35
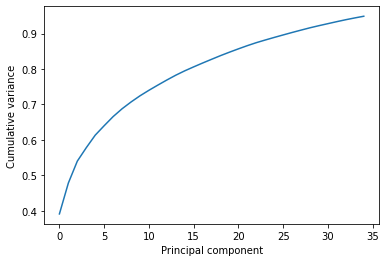
第35主成分まで含めると95%を超えることがわかりました。少しずつ増えていく感じです。
2-3-4. トラジェクトリの次元削減とプロット
PCAを使ってトラジェクトリを次元削減してみましょう。transform()を使うと、指定した数(n_components)の主成分に変換されます。

戻り値はサイズ「(トラジェクトリのフレーム数, 主成分の数)」のndarrayです。
主成分の数を3としてCα原子について変換するとこんな感じです。
# CαについてPCAで次元削減 transformed = Ca_pca.transform(md_universe.select_atoms("name CA"), n_components=3) print("shape of the array : ", transformed.shape) # shape of the array : (50, 3)
2次元にプロットして確認してみましょう。準備としてPandasのデータフレームに変換します。
import pandas as pd # トラジェクトリの1フレームの時間(ps) frame_time = md_universe.trajectory.dt # データフレームに格納する df = pd.DataFrame(transformed, columns=['PC1', 'PC2', 'PC3']) df['Time (ps)'] = df.index * frame_time df.head()
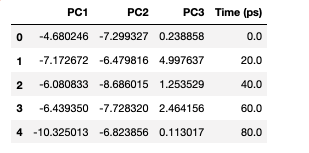
20ps 毎の各フレームで、主成分PC1〜PC3の重みに変換されました。
プロットしてみます。時間推移( Time (ps))で色づけしています。
import matplotlib.pyplot as plt import seaborn as sns pair_grid_plot = sns.PairGrid(df, hue='Time (ps)') pair_grid_plot.map(plt.scatter, marker='.')

PC1とPC2の2次元プロットでは近い色の点が集まった意味ありげなパターンになりました。
・・・何の意味かはわかりません。
2-3-5. 主成分の動きを動画で
PCAを使ったトラジェクトリの基本的な動きの確認には、プロット以外にも動画で確認する方法もあるそうです。
元々のトラジェクトリを各主成分に投影します。
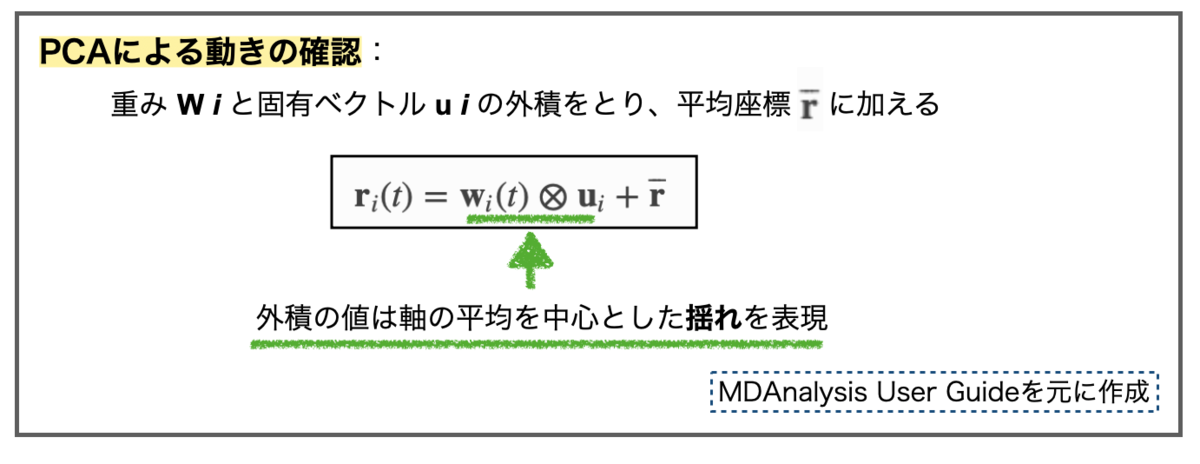
重み(w)は先のtransform()で得られていて、主成分の固有ベクトルはresults.p_components、平均座標はmeanでアクセスできます。上記式を実行しましょう。
# 第1主成分 pc_1 = Ca_pca.results.p_components[:, 0] # 第1主成分についての重み weight_1 = transformed[:, 0] # 平均座標 mean_coord = Ca_pca.mean # 投影 projected_1 = np.outer(weight_1, pc_1) + mean_coord.flatten() # arrayの形を整える coordinates = projected_1.reshape(len(weight_1), -1, 3) print(coordinates.shape) # (50, 317, 3)
最後のreshapeでは、投影を計算後のarrayを「フレーム数、原子数(N)、xyz座標(3)」の次元となるように整えています。
各原子の座標の時間(フレーム)変化のデータが手に入ったので、これをもとにUniverseオブジェクトを作成します。
proj_univ = mda.Merge(md_universe.select_atoms("name CA")) proj_univ.load_new(coordinates, order="fac") # <Universe with 317 atoms>
Merge()で既存のAtomGroupから新しいUniverseオブジェクトを作り、load_newで座標の情報を更新しています。
NGLviewを使ってトラジェクトリの動画を確認しましょう。
import nglview as nv view = nv.show_mdanalysis(proj_univ.atoms) view
全体が上下方向にクシャッととつぶれるような動きをピクピクしています。
ちなみにPCAで次元削減するまえの元々の動画はこんな感じです。
view = nv.show_mdanalysis(md_universe) view
そのまま見ているよりもPCAの方が動きがシンプルでわかりやすいですね。
PCAのプロットで第1主成分とパターンをつっくていた第2主成分ではどうなるでしょうか?(コードは同じなので省略します。)
今度は左右方向に揺れている感じです。第1主成分と第2主成分で異なる動きの向きが取り出せていそうです。
楽しい!!・・・意味はわからないですが。
主成分分析は以上です。
3. 相互相関解析(cross correlation)
3-1. 相互相関?
続いて相互相関解析を行います。こういう感じの話です。

こちらも動きの解析ですが、PCAでは基本的な運動のモードを取り出す感じだったのに対し、分子内の運動の相関に焦点を当てているようです。
3-2. PyTrajのインストール
MDAnalysisにそれらしい項目がなかったので、Making it rainで解析に使われていた PyTraj を利用したいと思います。
PyTrajはAMBERのAmber Toolsに組み込まれているcpptrajというプログラムをPythonから扱えるようにしたフロントエンドパッケージだそうです
- GitHub:PyTraj
- ドキュメント:pytraj 2.0.2.dev0
インストールはcondaでできます(チャンネルはambermdです)*2。
conda install -c ambermd pytraj
ただし上記はPython バージョン3.9以上では使えません。Python 3.8以下の仮想環境を作るか、AmberToolsを丸ごとインストールすればPyTrajを使えます*3。
AmberToolsのインストールもcondaでOKです。
conda install -c conda-forge ambertools=21 compilers
こっちはPython 3.9の環境にも入りました。
3-3. PyTrajでトラジェクトリファイルの読み込み
PyTrajで解析を行うためにトラジェクトリを読み込みます。
load()にトラジェクトリファイル(ex. DCD)とトポロジーファイル(ex. PDB)を渡します*4。
import pytraj as pt traj = pt.load("simulation_data/trajectory.dcd", "simulation_data/3poz-processed.pdb") print(traj) # pytraj.Trajectory, 50 frames: # Size: 0.064427 (GB) # <Topology: 57648 atoms, 17895 residues, 17881 mols, PBC with box type = cubic>
なお、PyTrajには他の解析ソフトとの相互利用を意識した機能もあり、MDAnalysisのUniverseオブジェクトからトラジェクトリを読み込むこともできます(PyTraj tutorial:Interface with mdanalysis)。
# MDAnalysisのUniverseオブジェクトから座標をnumpyのarrayに取り出す。 all_atoms = md_universe.select_atoms("all") coords_from_mda = np.asarray([all_atoms.positions for frame in md_universe.trajectory], dtype='f8') # 座標とPDBファイルからPyTrajに読み込む traj_from_mda = pt.Trajectory(xyz=coords_from_mda, top= "simulation_data/3poz-processed.pdb") print(traj_from_mda) # pytraj.Trajectory, 50 frames: # Size: 0.064427 (GB) # <Topology: 57648 atoms, 17895 residues, 17881 mols, non-PBC>
こちらはなぜか周期境界条件が認識されず、non-PBCとなってしまいました。原子数やトラジェクトリのフレーム数など合っていそうです。
3-4. PyTrajでアラインメント
ファイルから読み込んだトラジェクトリでアラインメントをとっておきます。
align()を使います。デフォルトでは全ての原子について最初のフレームを参照構造(ref)にしてアラインメントを取ります。
PyTraj(というよりAmber?)では、原子の選択はmaskと呼ばれるようです(Amber mask selection)。特定の原子団を選択したければ「mask=選択基準」といった形式で引数に渡すことになります。
例えば、Cα原子についてアラインメントを取るには引数mask='@CA'を渡します。
# Cα原子について最初のフレームを参照にアラインメントを取る traj_align = pt.align(traj, mask='@CA', ref=0)
無事、アラインメントが取れました。
3-5. PyTrajで相互相関解析
PyTrajでの解析準備が終わったので相互相関解析を行います。
Matrixのcorrel()を使うことで相互相関の行列が得られます。Cα原子を選択して計算しましょう。
from pytraj import matrix mat_cc = matrix.correl(traj_align, mask ='@CA') print(type(mat_cc)) print(mat_cc.shape) # <class 'numpy.ndarray'> # (317, 317)
計算対象のタンパク質構造の残基数が317なので、「アミノ酸残基 x アミノ酸残基」の2次元行列が得られました。
あとはこれをプロットするだけです。seaborbでヒートマップを可視化します。
ax = sns.heatmap(mat_cc, vmin = -1, vmax = 1, cmap = 'PiYG_r', square=True, xticklabels=30, yticklabels=30) ax.set_xlabel('Residues') ax.set_ylabel('Residues')

① 60 ~ 90(残基番号 760 ~ 790)、② 120 ~ 160(残基番号 820 ~ 860)、③ 180 ~ 240(残基番号 880 ~ 940)あたりに相関が大きくなっている群がありそうです。
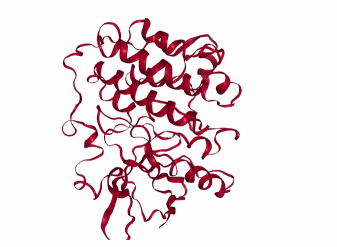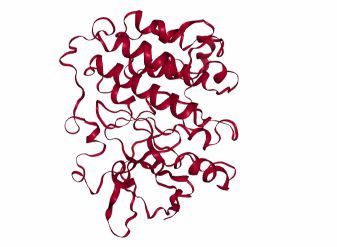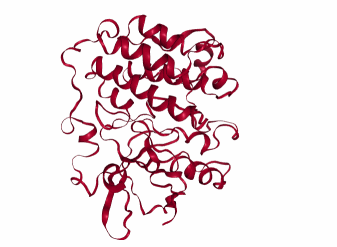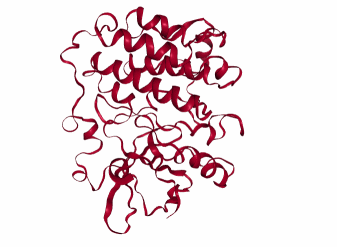
PyMolで少し色分けしてみました。

ヘリックスだったりシート構造だったりする部分のようです。「協同運動している箇所」と言われると「そうかも・・・???」という感じですが雑過ぎですね。。。ごめんなさい。
4. おわりに
以上、今回の記事ではMDトラジェクトリの解析(後半戦)を行いました。取り上げた解析は主成分分析と相互相関解析で、MDAnalysisとPyTrajを利用しました。
どちらも揺らぎの大きな特徴をつかむ解析のようですが、主成分分析では運動モードの種類を取り出す感じで、相互相関解析では分子内のアミノ酸残基の協同運動の傾向がでる感じでした。
個人的には第1主成分と第2主成分でそれぞれに投影した動画の運動の向きが変わっていたのが面白かったです。
短いシミュレーションでデフォルトの解析を実行しただけなので生物学的に意味のあることを言えるような記事ではありませんが、とりあえずの使い方がわかったのでヨシ!
以上、今回も間違いが多そうです。ご指摘いただければ幸いです。
ではでは!