OpenMMをステップバイステップで 〜 Part 1:GUIでタンパク質の前処理 〜
MD計算について調べてみるとオープンソースツールがたくさんあることがわかってきました。ここではGUIが用意されていて、初学者に優しそうなOpenMMでちょっとずつ遊んでみたいと思います。
この記事では第一歩、PDBから取得したタンパク質構造をMD計算にあわせて前処理する方法を試してみます。具体的にはOpenMM SetupでGUIによる処理をおこないます。次回の記事で、同じ処理をPython経由で行う方法をPDBFixerでためします。*1

1. この記事の目標
最後の最後にやりたいことはTeachOpenCADDのT019 · Molecular dynamics simulationとT020 · Analyzing molecular dynamics simulations で行われているようなタンパク質-リガンド複合体のMDシミュレーションと結果の解析を理解することです。
TeachOpenCADDはとても分かりやすい教材ですが、私のようなレベルの低い初学者にはそれでも少し要素が多すぎてついていけないです。もっともっと初歩の初歩から・・・
ということで、まずはMD計算に合わせたタンパク質構造の前処理を試します。TeachOpenCADDと同じ構造(PDB id: 3POZ)を題材にしますが、ここではタンパク質のみでリガンドは考慮しません。
この記事では、まずOpenMMに付随するGUIツールのOpenMM Setupで前処理の流れを確認します。
2. 準備
この記事で使うツールはOpennMMとOpenMM Setupです。すべてcondaがあれば簡単にインストールできます。
conda install -c conda-forge openmm conda install -c conda-forge openmm-setup
計算対象の構造は「タンパク質EGFRのキナーゼドメインとリガンドTAK-285の複合体(PDB ID: 3POZ)」です。

出典となっている論文はこちら
Structural Analysis of the Mechanism of Inhibition and Allosteric Activation of the Kinase Domain of HER2 Protein. Aertgeerts, K., Skene, R., Yano, J., et. al., (2011) J Biol Chem 286: 18756-18765 DOI: 10.1074/jbc.M110.206193
Open Access, CC BY 4.0
3. どうして前処理が必要なの?
そもそもどうしてPDBの構造をそのままシミュレーションにかけるのは要注意なのでしょうか?PDBFixerのマニュアルのイントロダクションが分かりやすいです。

このような問題に対してPDBFixerでは以下のような処理が可能になっています。

これからOpenMM SetupをつかったPDBの修正方法をみていきますが、GitHub : openmm-setupをご覧いただけばわかる通り、裏側ではPDBFixerが走っています。
4. OpenMM Setup で前処理しよう
それでは早速、OpenMM Setupを使った前処理をためしてみましょう!
処理の全体の流れは以下です。合わせて処理の前後の構造を描画しています(PyMol)。
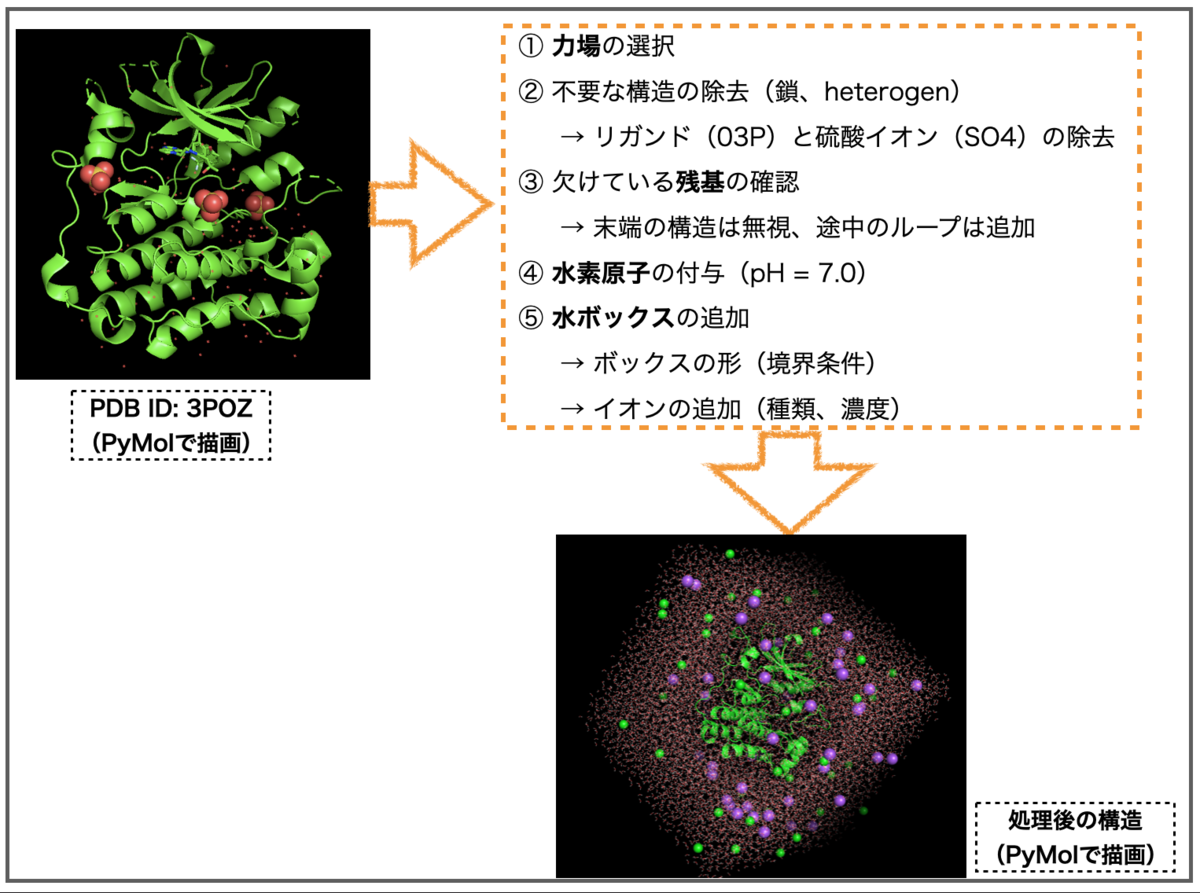
4-1. アプリの起動とファイルの読み込み
OpenMM Setupアプリをcondaでインストールした場合は、仮想環境をアクティベートして以下のコマンドでブラウザが立ち上がります。*3
openmm-setup

PDBファイルを扱うのでそのままcontinueします。
次の画面では① PDBファイルの読み込みと、② 力場の選択、③ 水モデルの選択を行います。Browseをクリックして入力ファイル(Input File)を選択できます。

力場(Force Field)と水モデル(Water Model)はプルダウンから選べます。選択肢を上図右側に貼りました。
ここではTeachOpenCADDに合わせてAMBER14とTIP3P-FBを選択しています。違いはよくわかりません。。。
上図下部では「入力ファイルに修正が必要か否か?」を聞かれます。Yesを選んで修正を加えていきましょう!
4-2. 不要な構造の除去
ついでPDBファイルの構造からシミュレーションの対象にしたい部分を取り出します。複数の鎖(chain)が含まれる場合やリガンド・塩などが入っている場合があるので、不要な構造を除去します。
以下のような画面で設定します。右側の3Dモデルはインタラクティブ表示で、ぐりぐり回転したりズームしたりできます。便利!
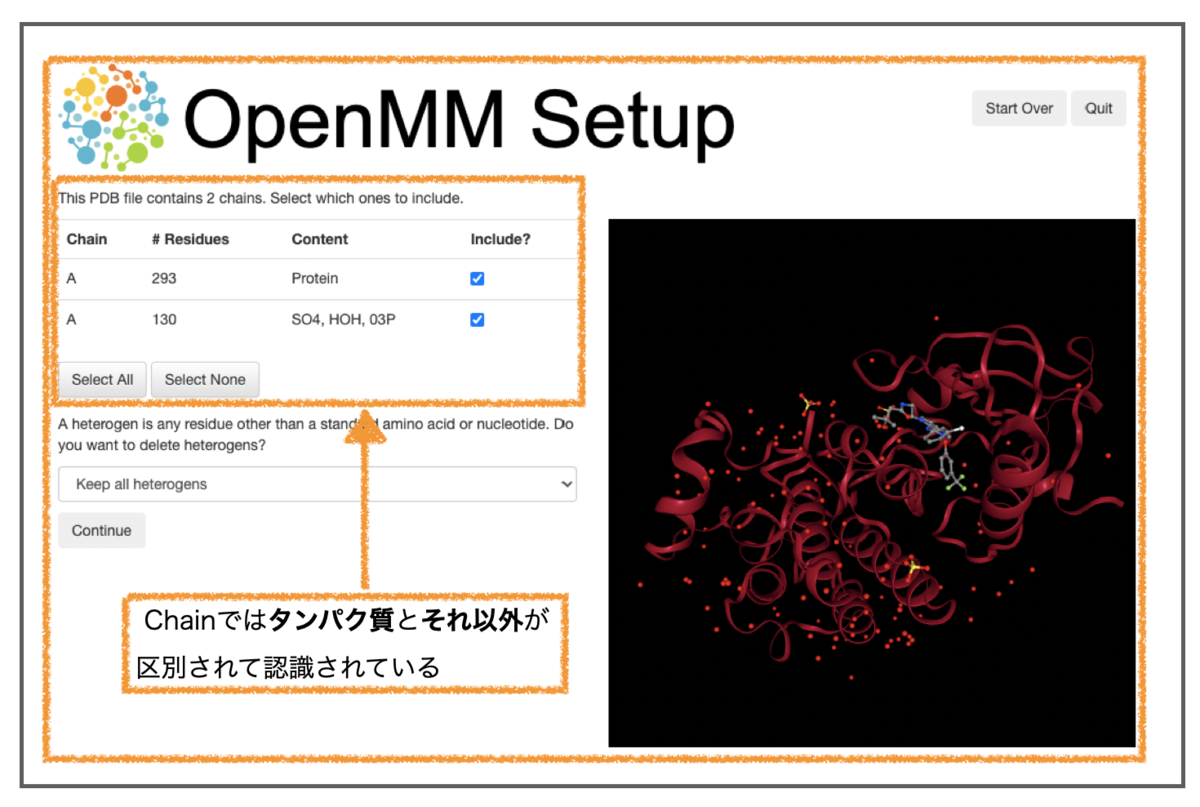
左側上部では鎖の選択ができます。Contentの部分を見るとProteinとそれ以外(SO4、HOH、03P)が区別されているのがわかります。テーブル右端のincludeのチェックの有無で構造を含めるかどうか選べます。
今回のシミュレーションでは硫酸イオン(SO4)とリガンド(03P)を除いた、タンパク質と水(HOH)を対象とします。上部テーブルではProtein以外の分子を水を含めてまとめてあつかっているため、チェックを外すとHOHも消えてしまいます。
そこで上部はこのままにして、下部のHeterogenの設定箇所を使います。

上図のようにDelete heterogens except waterを選択します。これで水を残したまま、それ以外のイオン、リガンドを削除できます。設定の変更は右側3Dビューにも反映され、リガンドなどが消えていることがわかります。
確認できたらContinue!!
4-3. 欠けている残基の確認と追加
続いてタンパク質構造で欠けている残基(missing residue)の確認を行います。以下のように残基の位置と種類を確認することができます。

残基を追加したい時は右端のAdd?にチェックを入れます。今回は両末端は加えず、途中の欠けている残基は追加することにします。(鎖が途中で切れたままだと変な動きになりそうなので・・・)
UniProt KB:P00533(EGFR_HUMAN)等と比較すればわかりますが、「PDB ID:3POZ」のカバーする残基番号は「696 - 1022」です。なので上図でチェックを入れていない「696 to 700」「1018 to 1022」がN末端とC末端に相当します。
参考までに欠けている残基の箇所を確認してみましょう。RSCB PDBサイトの3D Protein Feature Viewが非常にわかりやすいです。

上図左側の「UNMODELED」とがシークエンスの中でモデル化されていない部分です。missing residueはここを見れば良さそうです。
右側の3Dビューでピンク色にハイライトされたシート構造はUNMODELEDな領域に挟まれた2次構造(secondary structure)です。このシート構造につながる箇所が点線で表示されています(水色の矢印)。残基が欠けている部分は点線で仮につながれているようですね。
3D構造とシークエンス表示を対応させてみた感じループやタンパク質の両末端といった箇所が欠けているように思われます。フレキシブルで電子密度がうまく観察できなかったのでしょうか?
なお、上の「3D Protein Feature View」は以下のようにたどると見られます。

4-4. 重原子の欠けた残基の確認
「欠けている残基」につづいて、見えている残基の中で「一部の重原子が欠けているもの」の確認画面に遷移します。
以下の通り、1017番目のロイシン(Leu)残基でOXT原子が欠けています。

OXTはC末端の酸素原子(の負電荷を帯びた状態)を表す標準的な名前だそうです。一つ前のステップで確認した通り、この構造では1018番目以降の残基が欠けています。(本来の)C末端の構造が見えていないため、1017番がPDBファイル末端構造になっているようです。
この画面は「欠けている原子を足すよ!」っていうだけなので、このままContinueして良さそうです。
4-5. 水素原子の付与
ついで、水素原子を付与します。どのようなpH条件の設定の下で水素原子を付与するか選べます。

今回は中性条件での状態としたいので、上のように「pH = 7.0」としました。アミノ酸残基には酸性、中性、塩基性といろいろな官能基がありますが、設定したpH条件下の適切な状態にそれぞれ自動的に水素原子を付与してくれるようです。
つづいて同じ画面で水ボックスの追加を行います。Continueを押さずに次項をご覧ください。
4-6. 水ボックス(+イオン)の設定
最後に水ボックスの設定を行います。先の項で見た画面の下部「Add Water/Membrane」でAdd water boxにチェックを入れると下図のように設定画面が出てきます。

まず、水ボックスのサイズと形を設定します。今回は「タンパク質構造を取り囲むように水の層を追加(Padding)」します。
上図のようにSpecify a padding distanceを選択し、「Padding Distance (nm): 1.0」、「Box Shape:Cube」としました。これで厚さ1.0 nmの水の層で、全体が立方体(Cube)状になるようにタンパク質を囲む水ボックスができることになります。
Cube以外のボックスの形としてはTruncated octahedron、Rhombic dodecahedronといったものがあります。(境界条件に関係する設定項目のようですがどれがいいかよくわかりません。)
次に、イオンを追加します。イオン濃度(Ionic strength (molar))と、イオンの種類(Positive ion、Negative ion)を選びます。上図では0.15 Mの濃度でNaClを追加しています。他に利用可能なイオンはプルダウンの中身でご確認ください。
以上で前処理の設定は全て終わりです!Continueを押してください。
4-7. 処理後のPDBファイルの保存
PDBファイルの前処理が全て終わると、そのままシミュレーション条件の設定画面に遷移します。この記事ではこの先までは進みません。
下図のような画面になるのでSave Processed PDB File を押して前処理が終わった状態のPDBファイルを保存することができます。

3poz-processed.pdbという名前のファイルが保存されました。PyMolで描画すると以下のようでした。

前処理の途中で欠けている残基のうち鎖の途中の箇所は追加しましたがどのようになっているでしょうか?処理前後の構造を重ねて表示してみました。グリーンが処理前(3poz.pdb)、ブルーが処理後(3poz-processed.pdb)です。*4

上図の通り、処理後は残基が追加され実線で表示されています。PDBFixerの機能の一つに「欠けているループの構築」があげられていましたが確かに実行されていますね!
5. おわりに
以上、今回はMD計算を行う前のタンパク質の処理について試してみました。
OpenMMの提供するGUIツール OpenMM SetupのPDBファイル前処理機能を利用しました。指示に従ってボタンを押していくだけで使えてわかりやすいですね!
処理に必要な項目と操作の流れが大体わかったので、次回、同じ操作をPythonからPDBFixerを使って実行する方法を試したいと思います。
計算関係は前処理が大事とよく言われますが、具体的にどうすれば良いかわからないので難しいです。こればっかりは計算対象ごとの各論なので仕方ないのかもしれません。*5
今回も色々と間違いが多そうです。ご指摘いただければ幸いです。
ではでは!