OpenMMをステップバイステップで 〜 Part 2 PDBFixerでタンパク質の前処理 〜
前回の記事でMD計算に向けたタンパク質構造の前処理について、OpenMMに付随するGUIツールOpenMM Setupを使った操作を試しました。GUIは操作の流れを把握する上では便利ですが、処理を自動化することを考えるとコマンドやプログラムで扱えた方が良さそうです。
OpenMM SetupではPDB構造の修正に主にPDBFixerが使われています。また、TeachOpenCADDではPDBFixerのPython APIをつかうことで前処理を行なっています*1。
そこで今回はJupyter notebook上でPDBFixerを使って前回の操作をたどってみます。といってもTeachOpenCADDのT019 · Molecular dynamics simulationからコードを切り貼りさせていただくだけですけどね!
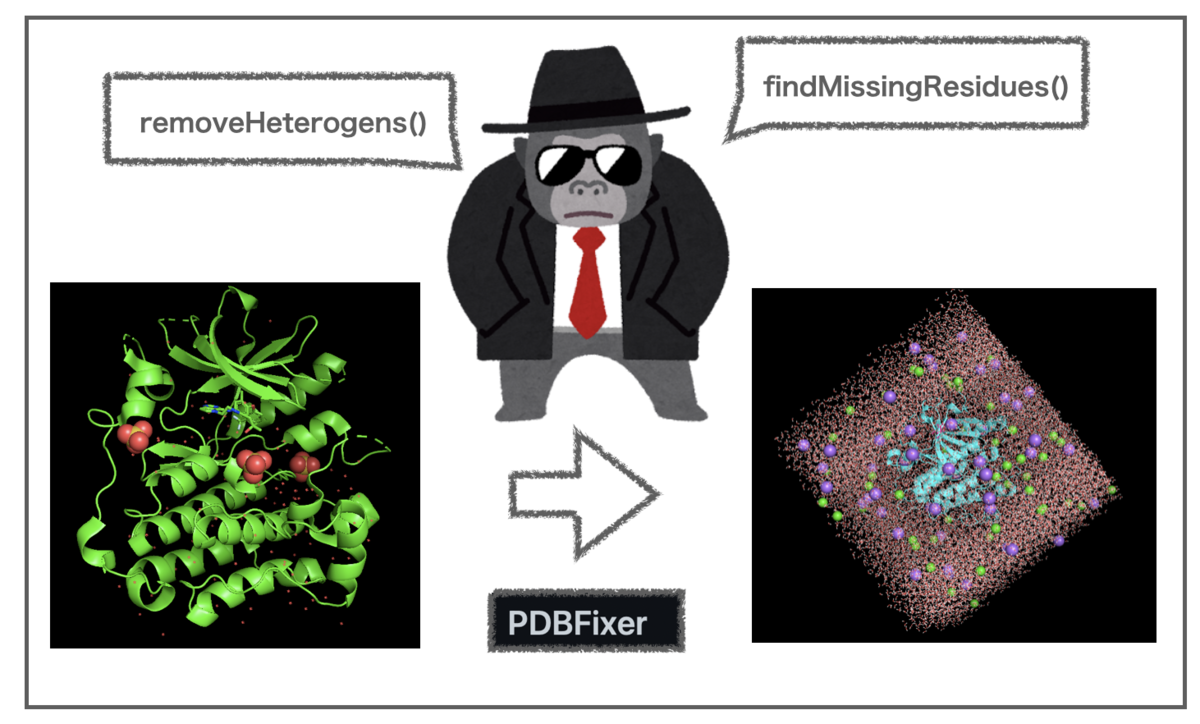
1. 処理の流れの確認
まずは処理の全体を再確認します。対象とするタンパク質の構造はPDB ID: 3POZで、以下のような処理を行いました。
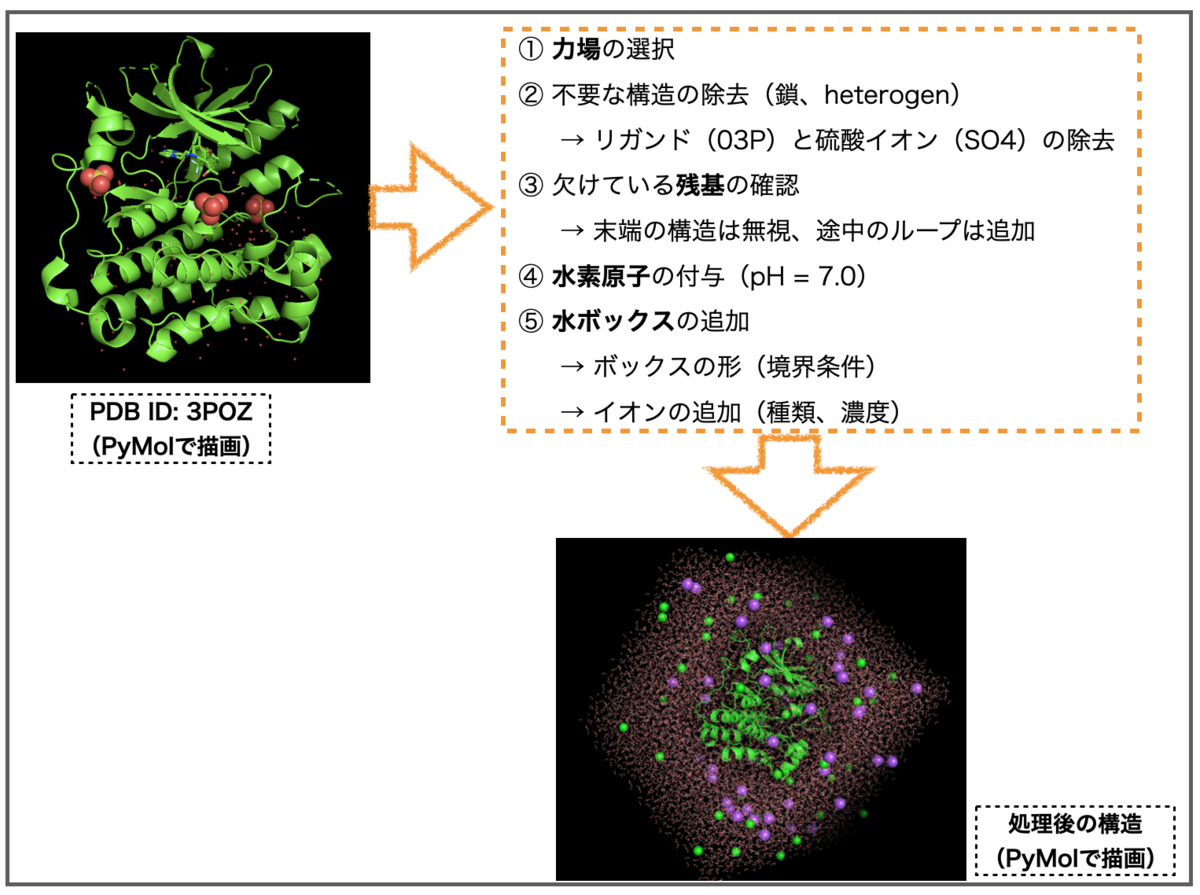
以上の操作をPythonベースで再現します。
2. PDBFixerで前処理を再現しよう
2-1. 準備
今回使うツールはOpenMMとPDBFixerです。PDBFixerもcondaでインストールできます。
conda install -c conda-forge pdbfixer
- GitHub : PDBFixer
- マニュアル:PDBFixer manual
タンパク質構造の処理のメインはPDBFixerですが、分子力場の設定や水ボックスの配置、またPDBファイルの出力といった操作でOpenMMを利用することになります。*3
2-2. 処理の全体
最初に処理全体のコードをまとめておきます。TeachOpenCADDのT019から抜粋して先に見た流れに合うように構成しています。*4
# 処理したいPDBファイルの指定 pdb_file = 'Data/3poz.pdb' # 必要なライブラリのインポート import openmm as mm import openmm.app as app from openmm import unit import pdbfixer # PDBFixerでPDBファイルを読み込む fixer = pdbfixer.PDBFixer(pdb_file) # 分子力場の設定 forcefield = app.ForceField("amber14-all.xml", "amber14/tip3pfb.xml") # 不要な構造の削除 fixer.removeHeterogens() # 欠けている残基のチェック(欠損原子の確認のためにも必要) fixer.findMissingResidues() # タンパク質末端の欠けている残基を取り除く処理 chains = list(fixer.topology.chains()) keys = fixer.missingResidues.keys() for key in list(keys): chain = chains[key[0]] if key[1] == 0 or key[1] == len(list(chain.residues())): del fixer.missingResidues[key] # 非標準な残基が含まれているか確認、あれば標準的なものに置き換える fixer.findNonstandardResidues() fixer.replaceNonstandardResidues() # 欠けている原子の確認、あれば追加する fixer.findMissingAtoms() fixer.addMissingAtoms() # 水素原子の付与(pHを設定する) ph = 7.0 fixer.addMissingHydrogens(ph) # 水ボックスの追加(力場、paddingの厚み、イオン濃度(デフォルトはNaCl)) modeller = app.Modeller(fixer.topology, fixer.positions) modeller.addSolvent(forcefield, padding=1.0 * unit.nanometers, ionicStrength=0.15 * unit.molar) # 処理後の状態(トポロジー、原子の位置)をPDBファイルで出力 top = modeller.getTopology() pos = modeller.getPositions() app.PDBFile.writeFile(top, pos, open('Data/processed.pdb', 'w'))
上のコードを実行することで、作業ディレクトリのData/3poz.pdbファイルを読み込んで処理した後、同じDataディレクトリに処理後のPDB(processed.pdb)が書き出されます。
PyMolで見た感じ再現できてそうです!
なお、追加したイオン(Na+、Cl-)の位置は毎回変わることはご了承ください。*5
今回の記事の要点は以上で、あとは一つずつ中身を見ていくだけです。
3. ひとつずつ処理を確認
では、順番に操作の内容を確認してみましょう!
PDBFixerはデスクトップアプリやコマンドラインアプリとしても使えるようですが、Python APIとして使うのが最も機能を有効利用できるらしく、マニュアルでもおすすめされています。
3-1. PDBFixerオブジェクト
PDBFixerをつかうには、処理したいPDBファイルを渡してPDBFixerオブジェクトを生成します。このオブジェクトに付随するメソッドを順番に呼んでいくことで、PDBファイルに対する処理が実行されていきます。
オブジェクトを作るのはこんな感じ。
fixer = pdbfixer.PDBFixer(pdb_file) print(type(fixer)) # <class 'pdbfixer.pdbfixer.PDBFixer'>
PDBFixerオブジェクトはトポロジー(topolgy)と位置(positions)の組み合わせという形で構造情報を保持しています。positionsはそのまま原子の位置です。topologyはもう一段上の抽象的な構造情報(ファイルに含まれるchain、各chainの残基、各残基の原子 etc.)といったものを指します。
試しにtopologyを表示してみます。こんな感じ。
print(fixer.topology) # <Topology; 2 chains, 423 residues, 2536 atoms, 2463 bonds>
構造全体を要約したような情報が出てきました。
同様にpositionsも確認できます。
print(fixer.positions[0:3]) # [Vec3(x=-0.032400000000000005, y=3.2759, z=-0.39270000000000005), Vec3(x=-0.0237, y=3.1343, z=-0.3461), Vec3(x=0.119, y=3.0783000000000005, z=-0.3514)] nm
多いので最初の3つだけ表示しています。リストのようなオブジェクトなのでスライスで切り出せます。
上記の通り(x, y, z)の3次元座標のベクトルで各原子の位置を表しているようです。参考までにPDBファイル(3poz.pdb)をテキストエディタで開いて一つ目の原子の情報を確認すると以下のようになっています(各フィールドに注釈を加えています)。
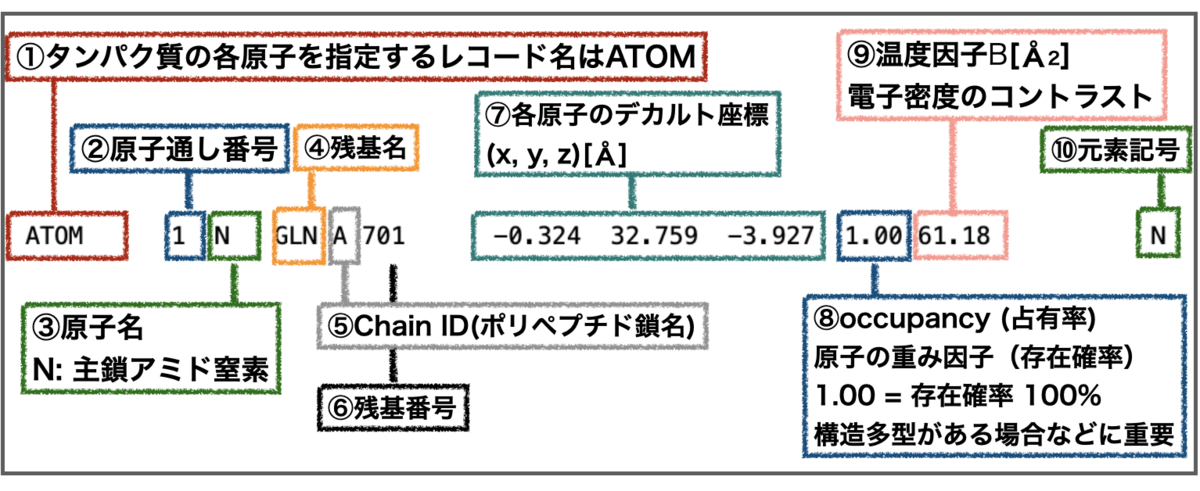
デカルト座標(⑦)の値を確認すると、PDBFixerオブジェクトのpositionsと一桁ずれています。PDBファイルはオングストローム(Å)、PDBFixerはナノメートル(nm)という単位の違いを踏まえれば一致していそうです。
PDBFixerオブジェクトがPDBファイル元にした構成であることがわかりました。*6
ちょっとくどくなりましたが、topologyとpositionで構造を表すという内容はOpenMMでも共通(?)で、処理後のPDBファイル出力の際にも関係してくるのでふれておきました。
3-2. 分子力場の設定
つづいて分子力場の設定です。「タンパク質構造の修正」という観点では関係なさそうですが、最後に「明示的な水分子を配置して水ボックスを作成する」という操作を行う上で必要になります。
ここではPDBFixerではなく、OpenMMのappに含まれるForceField() を使います。OpenMMでは力場の情報がXMLファイルで定義されており、それを指定して読み込むことで利用できます。*7
以下のように力場を設定します。
forcefield = app.ForceField("amber14-all.xml", "amber14/tip3pfb.xml")
引数の1つ目がメインの力場で、2つ目が水モデルを指定する力場です。
メインで使っているamber14-all.xmlはAMBER14力場の複数のファイルをまとめて読み込めるものです。種々の分子タイプ(amber14/protein.ff14SB.xml、amber14/DNA.OL15.xml、amber14/RNA.OL3.xml、amber14/lipid17.xml)をひっくるめて対応できて便利です。
水分子のモデルについてはamber14ディレクトリの中のamber14/tip3pfb.xmlを指定します。amber14ディレクトリの中のファイルは水だけでなく相当するイオンについての設定も含んでいるそうなので、このように力場を設定しておくほうが良いそうです*8。*9
3-3. 異質分子(heterogen)の除去
つづいてタンパク質以外の異質分子(heterogen)の除去です。「PDB ID : 3POZ」では硫酸イオン(SO4)や低分子リガンド(03P, TAK-285)といったものを除きつつ、水分子(HOH)は残すといった処理を行います。
以下の1行でおしまいです。
fixer.removeHeterogens()
PDBFixerのコード(pdbfixer.py)を参照するとわかりますが、デフォルトの引数ではkeepWater=Trueとなっているため、水分子(HOH)は残されることになります*10。
3-4. 欠けている残基の確認と除去(追加)
続いて欠けている残基(missing residue)の確認とそれらを「除去する or 追加する」を選択する操作です。ここで見つかったmissing residueは後ほど欠けている原子を追加する操作で補完されることになります。
以下のメソッドで欠けている残基を確認できます。
fixer.findMissingResidues()
見つかった欠損残基は以下のようにmissingResiduesで確認できます。(見やすさのためpprintで表示しています)*11
import pprint pprint.pprint(fixer.missingResidues) ''' {(0, 0): ['GLY', 'GLU', 'ALA', 'PRO', 'ASN'], (0, 33): ['GLU', 'GLY', 'GLU', 'LYS'], (0, 43): ['ARG', 'GLU', 'ALA', 'THR', 'SER', 'PRO', 'LYS'], (0, 156): ['GLU', 'TYR', 'HIS', 'ALA', 'GLU', 'GLY', 'GLY'], (0, 285): ['GLU', 'GLU', 'ASP', 'MET', 'ASP', 'ASP'], (0, 293): ['ILE', 'PRO', 'GLN', 'GLN', 'GLY']} ''' print('found ', len(fixer.missingResidues), 'missing regions') # found 6 missing regions
辞書型に格納されています。欠損残基として6つの領域が見つかりました。前回OpenMM Setupで確認した結果を再掲しておきます。
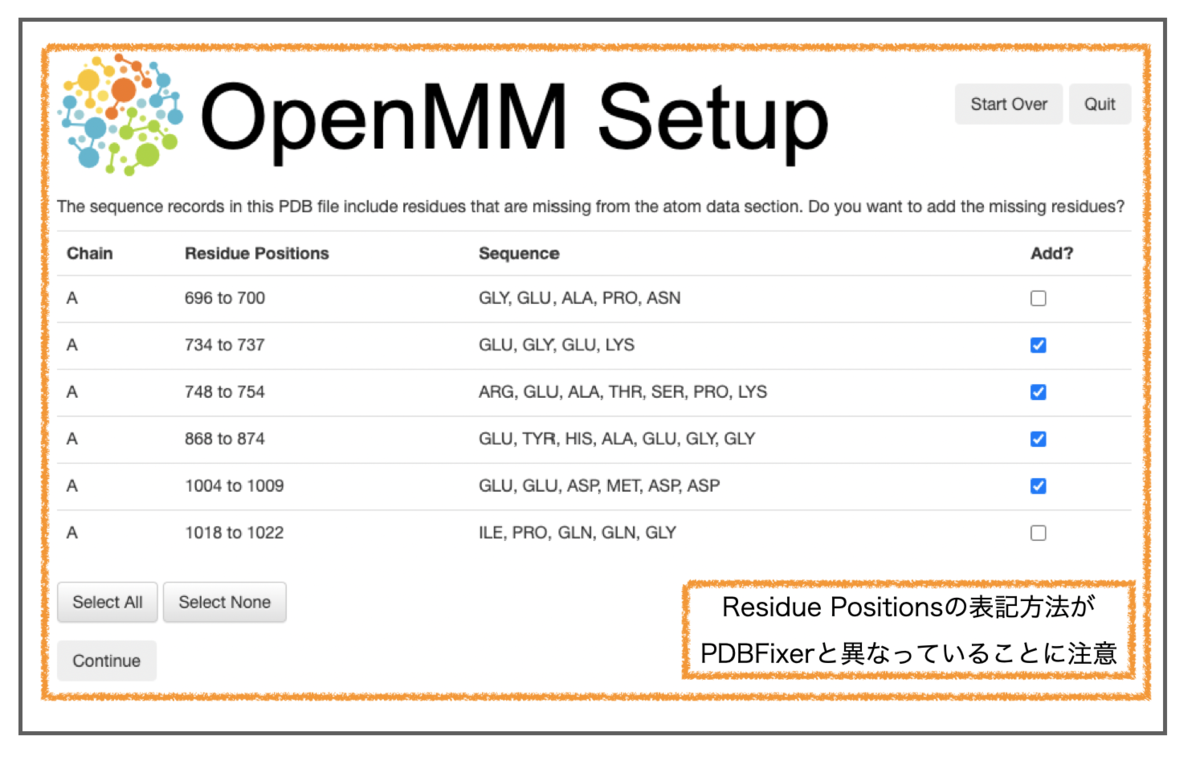
同じ結果が見つかっていそうです。辞書のKeyはタプル(chain.index, indexInChain)となっています。残基の位置は「鎖の中の位置(indexInChain)」で単純な残基番号(UniProtなどで確認できるもの)とは異なることにご注意ください。
見つかった欠損残基について、最終的な計算対象のモデル構造に含めたくない部分を取り除きます。タンパク質の両末端(N末端とC末端)を取り除き、鎖の中間部分(実験で見えなかったループ構造 etc.)を残したい場合は以下のようにします。
chains = list(fixer.topology.chains()) keys = fixer.missingResidues.keys() for key in list(keys): chain = chains[key[0]] if key[1] == 0 or key[1] == len(list(chain.residues())): del fixer.missingResidues[key]
先に見た通りmissing residueの辞書のKeyは(chain.index, indexInChain)でした。後者IndexInChainについて0(key[1] == 0)であれば欠損残基が鎖のN末端であることを意味します。逆に、鎖の残基数(len(list(chain.residues()))と一致した場合は鎖のC末端が欠損していることになります。これらに該当する箇所が見つかったら「delで削除する」というのが上記です。
この操作を行ったのちmissingResiduesは以下のように変化しています。
pprint.pprint(fixer.missingResidues) ''' {(0, 33): ['GLU', 'GLY', 'GLU', 'LYS'], (0, 43): ['ARG', 'GLU', 'ALA', 'THR', 'SER', 'PRO', 'LYS'], (0, 156): ['GLU', 'TYR', 'HIS', 'ALA', 'GLU', 'GLY', 'GLY'], (0, 285): ['GLU', 'GLU', 'ASP', 'MET', 'ASP', 'ASP']} ''' print('found ', len(fixer.missingResidues), 'missing regions') # found 4 missing regions
N末端とC末端の2つが消えて、辞書の中身は4つの領域となりました。
取り除かなかった欠損残基(この段階でmissingResiduesに残っているもの)は、次々項(3-6.)でaddMissingAtomsを行う際にPDBFixerが補完してくれるようです。ミッシングループの構築に相当する操作ですね。
なお、今回は行いませんでしたが、もし「全てのmissing residueを取り除きたい」場合は以下のように空の字辞書で置換してあげればOKです。
fixer.missingResidues = {}
3-5. 非標準な残基の確認と置換
つづいて非標準な残基を確認し、置換する操作です。これは前回の記事で確認したOpenMM Setup(GUI)の処理にはありませんでした。
今回対象としている 「3POZ.pdb」にはそもそも「非標準な残基」が含まれていないようなので、そのためかもしれません。一応該当するコードは以下です。
fixer.findNonstandardResidues() fixer.replaceNonstandardResidues()
NonstandarResiduesをfindしてreplaceする。PDBFixerのコードの文法がだいたいわかってきました。
なお、非標準なアミノ酸は結晶学上の目的で加えられたりするようです(結晶化しやすくしたりする?)*12。タンパク質結晶学は専門でないのでよくわからないです。
3-6. 欠けている原子の確認と追加
つづいて欠けている原子(missing atoms)の確認と追加を行います。前々項(3-5.)で行った欠損残基(missingResidues)の確認を踏まえた操作です。
まずは確認です。構造の中で欠けている重原子(heavy atom)を見つけます。
fixer.findMissingAtoms()
見つかった欠損原子は① missingAtomsと② missingTerminalsにわけて格納されます。
print(fixer.missingAtoms) # {} print(fixer.missingTerminals) # {<Residue 292 (LEU) of chain 0>: ['OXT']}
① missingAtomsは空で、② missingTerminalsには「292番 LeuのOXT」が格納されていました。
比較のため、前回OpenMM Setupで確認した結果を再掲しておきます。

同じ重原子が見つかっていますね!
では、欠けている要素を追加します。addMissingAtomsメソッドにより「① missingAtoms、②missingTerminals、③missingResidues」の3つのフィールドに格納された欠損要素が追加されます。((メソッドはAtomsとなっていますがResiduesもaddされるのがちょっとわかりにくいです))
fixer.addMissingAtoms()
引数にランダムシードを指定できることからわかるように、原子の追加の仕方(座標)にはランダムさがあります。
3-7. 水素原子の付与
さて、重原子について欠損の確認と追加が終わったので、今度は水素原子を付与します。この際pHを指定しますが、デフォルトの残基のpKaに従って水素原子の付与の仕方が決まるようになっています。
ph = 7.0
fixer.addMissingHydrogens(ph)
上では明示的に「pH = 7.0」の設定をしていますが、デフォルトが7.0なので空でも同じです。
3-8. 水ボックスの追加
最後にタンパク質構造を取り囲むように水ボックスを追加します。
OpenMMのappに含まれるModellerオブジェクトに変換した後で、addSolvent()で水分子を追加します。
# 水ボックスの追加(力場、paddingの厚み、イオン濃度(デフォルトはNaCl)) modeller = app.Modeller(fixer.topology, fixer.positions) modeller.addSolvent(forcefield, padding=1.0 * unit.nanometers, ionicStrength=0.15 * unit.molar)
先に見た通りPDBFixerオブジェクトはtopologyとpositionsで構造情報を保持しています。この2つを取り出してopenmm.app.Modeller()に渡すことでModellerオブジェクトに変換できます。
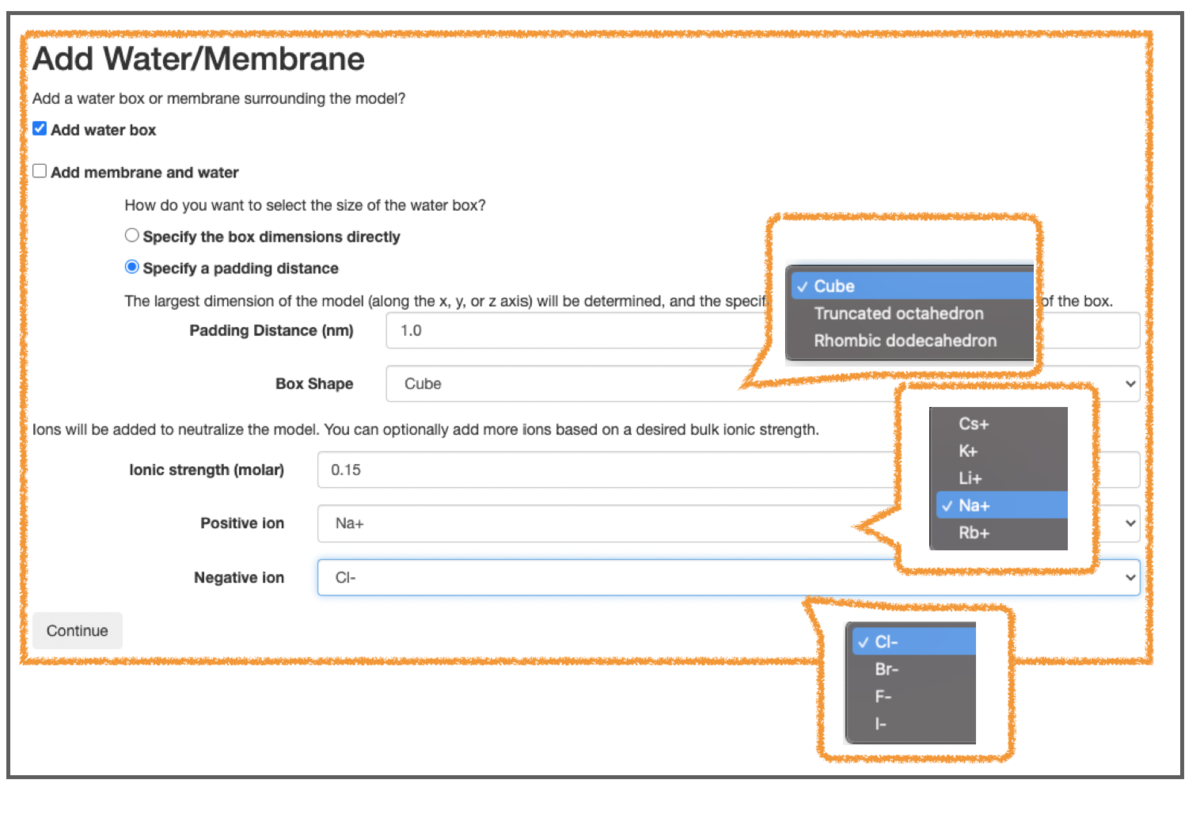
addSolvent()の引数は「力場(forcefield)、水の層の厚み(padding)、イオン濃度(ionicStrength)」などです。*13順番に見ていきますが、先にOpenMM Setupの該当箇所を再掲しておきます。
3-8-1. 分子力場
分子力場は先に「3-2. 分子力場の設定」で言及した通りです。addSolvent()ではデフォルトの水モデルが「TIP3P(model='tip3p')」となっています。力場の設定との互換性に注意したほうが良さそうです。
3-8-2. パディング
paddingではタンパク質を取り囲む水の層の厚みを指定します。
addSolvent()は基本的に「直方体の箱(rectangular box)に水を詰めていく」操作です。箱のサイズの指定方法には「① (x, y, z)ベクトルで各軸方向の長さを指定(boxVectors)」するか、「② タンパク質を中心としてその周りに水の層を厚さを指定して配置(padding)」するかの2通りあります。
「② padding」で指定する場合、タンパク質(溶質, solute)のxyz軸方向のうち一番大きい次元を求めて、その次元方向の両端にパディング距離分の厚みを加えた立方体の箱となります。つまり、1辺の長さ「最大次元 + パディング距離の2倍」の立方体です。
こういうイメージでしょうか?

3-8-3. イオン濃度と中和
ionicStrengthで指定した濃度となるようにイオンが配置されます(単位 = モル濃度, molar)。
デフォルトではイオンはNaClです。「positiveIon='Na+'」「negativeIon='Cl-'」といったように引数でイオンの種類を設定できるので必要に応じて変更します。
選択できるイオンはそれぞれ「‘Cs+’, ‘K+’, ‘Li+’, ‘Na+’, ‘Rb+’」「 ‘Cl-‘, ‘Br-‘, ‘F-‘, ‘I-‘」です。OpenMM Setupで見たものと同じですね。
ところでイオンの追加には系の中和も関係してきます。デフォルトではneutralize=Trueとなっているので、溶質が電荷を帯びている場合には、その電荷を打ち消してボックス全体の電荷の合計がゼロとなるようにイオンが追加されます。
なお、水ボックスへの配置の手順は以下の通りです。

上記の通り「ランダムに選んだ水分子をイオンと交換する」という配置の仕方なので、イオンの位置はランダムに変わります。
3-9. PDBファイルの出力
全ての処理が終わったので、最後に処理後の構造をPDBファイルで出力します。OpenMMのappにあるPDBFile.writeFile()を使えばOKです。
構造情報としてtopologyとpositions、それから出力先のファイル名を引数にわたしてやります。
# トポロジーと位置情報をそれぞれ取得 top = modeller.getTopology() pos = modeller.getPositions() # 出力先ファイル名とともに引数に渡して書き出す。 app.PDBFile.writeFile(top, pos, open('Data/processed.pdb', 'w'))
これでprocessed.pdbというファイルがDataディレクトリの中にできました。
おしまい!
3-10. (おまけ)+αの情報
PDBFixerの使い方は以上ですが、上記で触れなかったOpenMMの使用法に関する情報を追加します。
3-10-1. openmmのunitライブラリ
水ボックスを設定する際にナノメートルやモル濃度といった単位(unit)を利用しました。
ここでOpenMMのunitを利用しています。unitを使うと、数値とともにunit.nanometersやunit.molarを記述するだけで、通常の単位を記述するのと同じ書き方で単位込みの情報として扱うことができます。
untiはシミュレーションを設定する際に時間(フェムト秒、ピコ秒、ナノ秒)を記述する際にも使えるので、OpenMMを利用する上でおさえておいたほうが良さそうです。
3-10-2. PDBFixerのaddSolvent
今回はTeachOpenCADDの記述に合わせてModellerオブジェクトに変更したあとでaddSolvent()を実行しましたが、PDBFixerにも同じメソッドは組み込まれているので、オブジェクトの変更が無駄に見えるかもしれません。
PDBFixerのコードを見ると、実際の操作はOpenMMを利用していて、「①Modellerオブジェクトに一度変換、②addSolvent、③PDBFixerオブジェクトに戻す」ということが行われています。
ここでは、水ボックス作成後、そのままOpenMMでPDBファイルを出力して終わりですので、PDBFixerオブジェクトに戻す操作がない分、記事のように記載したほうが無駄が少ないかもしれません。
4. 終わりに
以上、今回の記事では前回OpenMM Setupで行ったタンパク質構造の前処理操作を、PDBFixerのPython APIで実行する方法を確認しました。
GUIで操作の流れを確認した後だと、だいたいやりたい処理の流れがわかるのがいいですね!
ひとつずつ処理を追ったせいで無駄に長くなってしまいました。毎度くどくてまとまりがなくてすみません。次回は、前処理済みの構造を使ったシミュレーションの設定方法を見ていきたいと思います。
今回も色々と間違いが多そうです。ご指摘いただければ幸いです。
ではでは!
*1:OpenMM Setupを利用していません
*2:イラストはいらすとやさんより
*3:PDBFixerは内部でOpenMMを利用しているので、OpenMMをインストールしていないと使えません。念のため・・・
*4:TeachOpenCADDはCC-BY-4.0ライセンスで公開してくださっています。感謝!
*5:後で確認しますが、設定した濃度(ionStrength)にあう数をランダムに配置しています
*6:尚、3poz.pdbファイルには異方性温度因子(ANISOU)の情報が各原子について記述されています。話が難しくなるので省略しています。
*7:力場についての詳細や利用できる力場の種類はドキュメントOpenMM User Guide : 3.6.2. Force Fieldsをご参照ください
*8:OpenMM User GuitdeのTIPの記載より
*9:尚、水分子の力場をTIP3P-FBとしていますが、このFBはForceBalanceの略のようです。元の文献はこちらです。(Lee-Ping Wang, Todd J. Martinez, and Vijay S. Pande Building Force Fields: An Automatic, Systematic, and Reproducible Approach J. Phys. Chem. Lett. 2014(5)1885DOI;10.1021/jz500737m)
*10:他に残される残基は、通常の残基とUNK残基などです
*11:参考: note.nkmk.meさんの記事「Pythonのpprintの使い方(リストや辞書を整形して出力)」
*12:PDBFixerのマニュアルの記述より
*13:引数の詳細はマニュアル addSolventを参照してください。