OpenMMをステップバイステップで 〜 Part 4-2 よくある質問(FAQ@GitHub)〜
OpenMMのシミュレーションを実行した前回の記事(part 4)で、結果の描画でタンパク質が境界から飛び出ているのが気持ち悪いと書きました。
OpennMMのGitHubページ「Wiki: Frequently Asked Questions」に全く同じ内容が書かれていました。全く気づいていませんでした。。。
よくある質問なら共有しておいた方が良いよね!ということで、DeepL翻訳(にして修正)したものを置いておきます。*1*2
- 1. 周期境界条件はどのように機能するの?
- 2.「粒子の位置がNaNです」「エネルギーがNaNです」といったエラーが出るのはなぜ?
- 3. 「残基のテンプレートが見つかりません」というエラーが出るのはなぜ?
- 4. SystemやForceに加えた変更が無視されるのはなぜ?
1. 周期境界条件はどのように機能するの?
非常によくある質問は、もとを探ると周期的境界条件の理解に関するものです。「なぜ水分子が周期境界条件の箱の外側に拡散するのか?」「なぜタンパク質が水のボックスの端からはみ出るのか?」といった質問がよくあります。 また、「特定の力が周期境界条件においてどのように適用されるか?(あるいは適用されないか?)」といったことに関する質問もあります。 一見、簡単な質問に見えますが、見た目よりもずっと複雑な問題なので、まず初めに説明しましょう。
概念的に言えば、周期境界条件でシミュレーションを行う場合、すべての「粒子」は、空間全体で繰り返しパターンをとる無限の粒子のグリッドを表します。 周期的なボックスが10nmの立方体で、ある粒子が(1, 2, 3)にあるとすると、(11, 2, 3)、(21, 2, 3)、(-9, 12, 103)・・・にも粒子があることになります。これらの位置のうち、どの位置をリストアップするかは重要ではありません。 どれをとっても、全く同じ粒子の無限のグリッドを表しています。
次に、理解すべき非常に重要なポイントがあります。
周期境界条件は、位置ではなく、変位量(displacement)に適用されます。
もう一度10nmの立方体を例にとりましょう。(1, 0, 0) と (9, 0, 0) の2つの粒子間の変位量はどうなるでしょうか? 周期境界条件がない場合、変位量は(8, 0, 0)となります、周期境界条件がある場合、(-2, 0, 0)となります。 もし、2番目の粒子の位置が(-1, 0, 0)や(99, 0, 0)であったとしても、それは問題ではありません。変位量は同じになります。 周期境界条件は、粒子間の変位量に依存する力を計算するときに適用されます。
もう一つ、非常に重要なポイントがあります。
周期境界条件は、非結合相互作用のみに適用され、結合相互作用には適用されません。
2つの粒子間の結合を考えてみましょう。それぞれの粒子は実際には無限の粒子のグリッドを表していますが、だからといって、最初の粒子のすべてのコピーが2番目の粒子のすべてのコピーに結合しているわけではありません。 最初の粒子の各コピーは、2番目の粒子の1つの特定のコピーと結合しており、それ以外のものは結合していません。 もし第2の粒子の異なるコピーが、結合しているコピーよりも何らかの方法で近づくことができたとしても、結合が突然「切れて」異なるコピーと結合をつくりなおすはずだということにはならないのです。
(実際には、本当に必要であれば、結合相互作用に周期境界条件を適用することは可能です。結合相互作用に対して setUsesPeriodicBoundaryConditions() を呼び出すだけでできます。 ですが、通常は行わないことで、特別な状況でのみ適切です。 例えば、一方の端がもう一方の端の次の周期的なコピーに結合しているような無限分子をシミュレートしたいといった場合があげられます。)
これは拘束条件や、実際には結合相互作用の一種となっている非結合相互作用の例外にも適用されます。非結合相互作用は通常、互いに結合している粒子間(1-2、1-3、1-4相互作用)では除外されるか、縮小されます。 これは直接結合している粒子にのみ適用され、その粒子の他の周期的なコピーには適用されません。
その結果、2つの異なる周期的なコピーの間で分子を壊してはいけないということになります。先に、粒子が(9, 0, 0)にあるか(-1, 0, 0)にあるかは重要でないと書きました。しかし、もしその粒子が(1, 0, 0)にある粒子と結合しているならば、それは重要なことなのです。 一方は結合長が8であることを示し、もう一方は結合長が2であることを示します。ほとんどのMDパッケージはこれと同じ要件を満たしているので、通常は問題ありません。 しかし、時々、すべての原子が独立して1つの周期ボックスにまとめられているファイルに出くわすことがあり、分子が真っ二つになっていることがあります。 MDTrajのmake_molecules_whole()関数など、このようなファイルを修正するためのツールがあります。
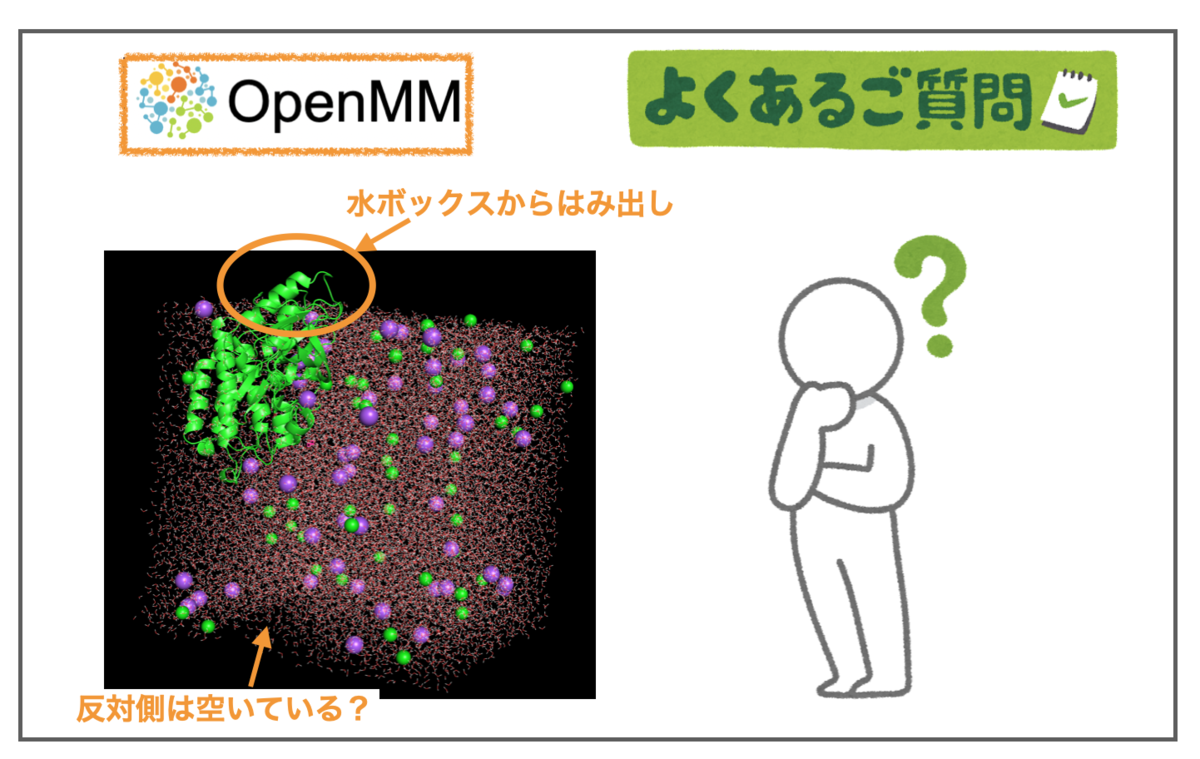
ここまではシミュレーションの実行に関することでしたが、次に出力ファイルの保存について説明します。 MDエンジンにとって、(0, 0, 0)を中心とする分子は(100, -30, -80)を中心とする分子と全く同一かもしれませんが、トラジェクトリを可視化しようとする場合、異なる分子が多くの異なる周期的な箱に散らばっていると何が起こっているか分からなくなってしまいます。 このため、Context.getState() には、返されたすべての粒子の位置を一つの周期的なボックスにまとめるオプションがあります。 同様に、レポーター関数(reporter)は周期系のトラジェクトリを書き出すときに自動的にこれを行います。ボックスにまとめる作業は常に分子全体に対して行われます。すべての分子の中心は1つのボックスの中にありますが、その分子のいくつかの原子はその外側にはみ出していることがあります。これが、タンパク質の一部だけが水に囲まれているように見えることがよくある理由です。
次のような重要なポイントがあるため、可視化はさらに複雑になっています。
一意に定義された「周期的なボックス 」は存在しない。
例えば、ボックスの幅が10である場合、粒子の位置はすべて0と10の間にあるべきということでしょうか? それとも-5から5の間でしょうか?どちらの方法も様々なプログラムで共通する慣習になっています。 タンパク質が原点を中心とする初期構造を読み込んで、すべての分子を0から10の間に置くようなトラジェクトリを書きだすと、タンパク質のほとんどが水の外に出てしまい、反対側の水のボックスに大きな穴が開いているように見えます。 これは非常によくある問題なので、多くの解析プログラムには特にこの問題に対処する機能が備わっています。例えば、MDTraj の image_molecules() 関数や Cpptraj の autoimage コマンドなどがあります。これらのツールは、すべての粒子を一つの箱に入れて、溶質を中心に、その周りに水を配置するような賢い方法をとります。
2.「粒子の位置がNaNです」「エネルギーがNaNです」といったエラーが出るのはなぜ?
NaNとは「not a number」の略で、「数字ではない」という意味です。NaNは、ゼロで割ったり、負の数の平方根を取ったりするような、実数値がうまく定義できない計算の結果をコンピュータが表現する方法です。このエラーが発生した場合、一般的にはシミュレーションが破綻(blown up)していることを意味します。このようなエラーは様々な理由で発生しますが、一般的には力や速度が大きすぎることが原因です。その結果、粒子が急激にジャンプし、次のタイムステップでさらに大きな力が発生し、数ステップですべての粒子の位置がNaNに変わってしまいます。
シミュレーションが破綻する原因には、さまざまな種類の問題があります。より一般的なものをいくつか紹介します。
タイムステップが大きすぎる場合。 タイムステップが大きすぎる場合、シミュレーションはしばらくの間、正常に実行されたように見えますが、その後、破綻します。タイムステップが大きすぎる場合、すぐに不安定になります。
初期コンフォメーションがエネルギー最小化されていない場合。 例えば、結晶構造を初期コンフォメーションとして使用している場合、座標が力場の最小値になっていない可能性があり、最初のタイムステップで非常に大きな力を発生させます。これは通常、局所的なエネルギー最小化を実行することで修正することができます。
周期的なボックスのベクトルが正しく設定されていない場合。 例えば、Amberのprmtopとinpcrdファイルから開始する場合によくある間違いです。どちらのファイルも周期的ボックスのベクトルを指定しており、お互いに異なる場合があります。粒子座標があるボックスサイズを想定していても、シミュレーションが異なるボックスサイズを使用している場合、粒子間の激しい衝突を引き起こす可能性があります。
拘束条件が正しく定義されていない場合。 拘束条件の適用には行列を逆行列にする作業が含まれます(明示的な場合もあれば、暗黙的な場合もあります)。この行列の条件が悪いと、シミュレーションが破綻する原因になります。単純な原因としては、同じ粒子ペアに2つの異なる拘束条件が課されている場合が挙げられます。また、同時にすべてを満たすことができない矛盾した拘束条件がある場合にも起こりえます。その他、より複雑な拘束条件のセットも、条件付けが悪い行列につながる可能性があります。
力の定義が正しくない場合。 これは、系(System)を直接構築しているとき、あるいは系に余分な力を追加しているときに、最も起こりやすい問題です。適用する力が大きすぎる場合、シミュレーションが破綻する可能性があります。また、カスタマイズした力を作成する際には、直接NaNを生成するような式を書かないように注意してください(負の値を取りうるの値の平方根を取る式など)。
このようなエラーは様々な原因で発生するため、デバッグが困難な場合があります。 最初に確認することは、シミュレーションがすぐに(最初の数ステップで)破綻するか、はじめは長時間実行されるかです。問題が初期コンフォメーション(最小化されていないなど)または系の定義(拘束条件や力など)のいずれかにある場合、通常すぐに破綻します。局所的なエネルギー最小化を実行してみて、それで解決するかどうかみてください。 一方で、ステップサイズが大きすぎる場合、はじめはしばらく実行されることがあります。その場合は、まずステップサイズを小さくしてみてください。
エラーが断続的なものではなく再現性がある場合、通常は何が起こっているのかを正確に追跡することができます。 まず、非常に大きな力がかかっている粒子を探すことから始めてください。そして、その力がどこから来ているのかを特定します。その力は、結合性相互作用によるものか、それとも非結合性相互作用によるものでしょうか? 具体的な相互作用を特定できますか? 例えば、2つの粒子が近すぎるのでは?
これらのエラーをデバッグする別のアプローチは、エラーの原因となるものを系統的に削除することです。力を1つずつ取り除いてみてください。 すべての拘束条件を取り除いてみる。あるいは、周期境界条件を無効にしてみる。少しの辛抱と忍耐で、大抵の場合、原因を特定することができます。
3. 「残基のテンプレートが見つかりません」というエラーが出るのはなぜ?
このエラーは、ForceField上で createSystem() を呼び出したときに発生することがあります。このエラーの意味を理解するためには、OpenMMで力場がどのように動作するかを少し知っておく必要があります。
力場を使って系(System)を構築する最初のステップは、すべての原子のタイプを特定することです。 原子のタイプは非常に固有のものである場合があります。例えば、全ての炭素原子が同じであるわけではありません。力場は、α炭素と芳香族側鎖の炭素とでは、おそらく異なるパラメータを使用するでしょう。 原子タイプは、残基をテンプレートにマッチングさせることで決定されます。力場は、認識できるすべてのタイプの残基のためのテンプレート定義を含んでいます。createSystem()を呼び出すと、渡されたトポロジー(Topology)のすべての残基をループして、それぞれをテンプレートにマッチングさせようとし、そのテンプレートで指定された原子タイプを割り当てます。
残基は2つの情報、すなわち各原子の元素とそれらをつなぐ結合(残基の原子と別の残基の原子をつなぐ結合も含む)の情報に基づいてテンプレートにマッチングされます。残基の名前など他の情報については気にしません。(ただし、後述するように、誤った残基名は問題を引き起こす可能性があります。) 元素と結合だけが重要です。
残基にマッチングするテンプレートを見つけることを妨げる多くの問題があります。ここでは、より一般的なものをいくつか紹介します。
力場のXMLファイルが省略されている。 これは最も単純な可能性の1つです。ForceFieldオブジェクトを作成し、どのファイルを読み込むかを指示したときに、単に1つだけ抜けている可能性があります。例えば、Amber14とCHARMM36の定義では、水を他の力場とは別のファイルに入れ、異なる水モデルを簡単に選択できるようにしています。そのファイルを指定しなかった場合、力場はトポロジー(Topology)の水分子に一致するテンプレートを持ちません。
非標準の残基がある。 AmberやCHARMMのような標準的な力場は、タンパク質や核酸のような様々な標準的な分子に対応しています。しかし、 Topologyにリガンドやその他の低分子が含まれている場合、 力場にはそのためのパラメータがない(つまり、 テンプレートがない)可能性があります。
トポロジーに原子がない。 例えば、 結晶構造には通常、水素原子が含まれておらず、 またフレキシブルな領域では重原子が欠落していることがよくあります。 これらの原子がない場合、トポロジーは力場のテンプレートとマッチングしません。 OpenMM-Setup は、この種の問題の多くを自動的に修正することができます。
水モデルが Topology と矛盾している。 4サイトおよび5サイトの水モデルは、各水分子が仮想サイトのための「余分な粒子」を含んでいることを想定しされています。これがない場合(あるいは逆に、あるのに3サイトの水モデルを使おうとした場合)、テンプレートがマッチングしません。
Modeller.addExtraParticles()を使って、この問題を修正することができます。鎖の終端が正しくない。 タンパク質、そして核酸はさらに、末端の構造が多種多様になっていることがあります。鎖の末端の残基は、中間位置の残基とは違っていて、異なるテンプレートが必要です。多くの場合、力場は各鎖の終端の構造について1つか2つしかサポートしていません。もしTopologyの末端が異なるものである場合、テンプレートにマッチングしません。
非標準のPDBファイルを持っている。 PDBは標準化されたフォーマットであり、specificationで正確に定義されています。 残念ながら、多くのPDBファイルはその仕様に従っておらず、様々な問題を引き起こしています。
最後のポイントについては議論が必要です。PDBファイルには様々な非標準の可能性があるからです。テンプレートは原子の元素と原子間の結合という2つの要素に基づいてマッチングされることを思い出してください。これらの一つでも不正確に再構築されると、テンプレートのマッチングでエラーが生じます。
PDBファイルの各ATOMレコードには、元素を指定するフィールドがあります。残念ながら、そのフィールドが空白のままのファイルもあります。その場合、PDBFileクラスは原子の名前から元素を推測しようとします。通常は正しく推測することができますが、常にそうとは限りません。
結合はより複雑です。PDB の仕様では、標準的な残基と異質原子(heterogen)を区別しています。標準的な残基(アミノ酸、ヌクレオチド、水)には結合が指定されていません。ファイルを読み込むソフトウェアが自動的に結合を追加することになっています。その他の分子はCONECTレコードで結合が明示的に指定されているはずです。これらのレコードがない場合、PDBを読むソフトウェアはどの原子が結合しているのかわからないので、ForceFieldはテンプレートをそれらにマッチングさせることができません。
標準残基には独自の問題があります。それらは名前によって認識されます。 PDB Chemical Components Dictionary ではすべての残基と原子の標準的な名前が指定されています。これにより、例えば、ALAという残基はCAという原子とCBという原子の間に結合があるはずだということがPDBを読むソフトウェアにはわかるわけです。残念ながら、ファイルによっては残基や原子の名前に非標準的なものを使っている場合があります。そのような場合、PDBFileはどこに結合を追加すればよいのかわかりません。
これが、原子や残基の名前が重要になる理由です。ForceFieldはテンプレートをマッチングするときに名前を使いません。しかし、ファイルが非標準的な名前を持っていると、PDBを読むソフトウェアは正しい結合を追加することができず、ForceFieldがテンプレートにマッチングすることができなくなります。
PDBFileは非標準的な名前に対応するように努めています。よく使われる非標準的な名前を認識するための大きなテーブルが用意されています例えば、水分子の標準的な名前は HOH ですが、それ以外にも WAT、SOL、TP3 などの名前も受け付けます。同様に、水分子の酸素の標準的な名前はOですが、OWやOH2という名前も受け付けます。これらの名前は、過去に様々なプログラムが書いたPDB ファイルで誤って使用したもので、OpenMM はそれらを探すことができます。しかし、OpenMMが認識できない他の非標準的な名前を使用している場合、結合は正しく追加されず、ForceField はテンプレートをマッチングさせることができなくなります。
4. SystemやForceに加えた変更が無視されるのはなぜ?
一旦Contextを作成すると、そのContextが作成されたSystemはイミュータブル(不変のオブジェクト)として扱われます。そのシステムに対して更に変更を加えても無視されます。これには、ForcesやVirtualSitesなど、Systemに含まれる他のオブジェクトも含まれます。
Contextの作成にはコストがかかります。GPU カーネルのコンパイル、スプラインの表形式関数へのフィッティング、拘束条件行列の構築と逆行列の計算など、シミュレーションの開始時に行う必要がある先行作業が多くあります。その過程で、Systemからの情報は多くの異なる場所にコピーされ、多くの場合、高度に処理された形でコピーされます。SystemまたはForceに変更が加えられると、多くの内部データ構造が無効になる可能性があります。そのため、OpenMM では、効率と混乱を避けるために、既存のContextはSystemに対するすべての変更を無視するというルールになっています。Contextを作成すると、必要なものはすべてSystemからコピーされ、それ以降は参照されません。
このルールは、状況によっては妨げになる場合があります。シミュレーションの過程で何かを変更することが本当に必要な場合があります。このような状況に対応するために、スピードと柔軟性のトレードオフの関係にある複数の方法が提供されています。
最初の方法はグローバルパラメータです。グローバルパラメータの値はSystemではなくContextに保存されるので、setParameter()を呼び出すことでいつでも変更することができます。カスタマイズしたフォースは全て、グローバルパラメータを定義することができます。 パラメータに依存する形でエネルギーを定義した場合、パラメータの新しい値を設定するだけで、いつでも簡単にエネルギーを変更することができます。 NonbondedForceでは、パラメータのオフセットで同様のことを行うことができます。グローバルパラメータを定義することで、粒子のセットを一度に協調的に変更することができます。
次に、ほとんどのフォースではupdateParametersInContext()メソッドが用意されています。これはSystemの限られた部分を更新するための制御された方法を提供します。例えば、粒子のセットの電荷を変更することができます。これを行うには、NonbondedForceに対してsetParticleParameters()を呼び出します。 他の変更と同様に、これは既存のContextでは無視されます。 しかし、次にupdateParametersInContext() を呼び出すと、変更されたパラメータ値が既存のContextにコピーされます。これは、グローバルなパラメータを変更するだけの方法よりもはるかに遅いですが、完全に新しいContextを作成するよりもはるかに速いです。この方法では、Forceのすべての条件を変更できるわけではないことに注意しましょう。updateParametersInContext() メソッドが何を更新し、何を更新しないかについては、各メソッドの API ドキュメントを参照してください。
最後の選択肢は、Context上でreinitialize()を呼び出すことです。 これにより、内部のデータ構造をすべて破棄し、一から作り直します。 これは、事実上まったく新しい Context を作成するのと同じことです。非常に遅いですが、絶対に全てを変更することができます。
内部データ構造がすべて破棄されるため、位置、速度、パラメータ値などの状態の情報も失われます。引数 reinitialize(preserveState=True)を渡すことで、それらの情報を保持するように要求することができます。これを実行すると、まずメモリ上にチェックポイントを作成し、次に Context を再初期化(reinitialize)し、最後にチェックポイントを再読み込みします。ほとんどの変更に対して、これは簡単に状態の情報を保持する方法を提供しますが、もしチェックポイントを有効でなくするような方法でSystemが変更された場合は、失敗して例外を発生させます。例えば、System内の粒子の数が変化した場合、チェックポイントをロードすることができなくなり、例外が発生し、状態の情報が失われます。
以上が、2022年4月現在のFAQです。