OpenMMをステップバイステップで 〜Part 6 MDAnalysisでトラジェクトリ解析(①)〜
OpenMMの使い方を順番にたどる記事のPart 6です。
前回まででOpenMMを使ったMDシミュレーションの準備&実行(とコードの書き方の理解)ができました。今回はシミュレーション結果のトラジェクトリ解析に挑戦したいと思います。
この記事で取り上げるソフトウェアはMDAnalysisです。またコードはTeach OpenCADD T20・Analyzing molecular dynamics simulationsを参考にしています。
途中で力尽きたので今回は解析のパート①です。*1

1. MD計算のデータ解析って何するの?
そもそもMD計算のデータ解析はどんなことをすれば良いのでしょうか?
私のような初学者にとって素晴らしい講演動画と資料がありました!
高度情報科学技術研究機構(RIST)のオンラインサロン 第4回 スパコンコロキウム でおこなわれた埼玉大学 松永 康佑先生のご講演です(研究室HP)。
発表資料(PDF):生体分子系の分子動力学シミュレーションデータの解析入門
①基本的な考え方から、②データ解析入門、③携わられたご研究の実例まで紹介してくださっていてとても楽しい講義です。
特に印象に残った点は「シミュレーション」(データの生成)と「データ解析」の「分業」に分野の研究スタイルが変化しているということです。専用のスパコンで計算したデータが公開され、別のグループがそのデータを使って新規な解析手法の論文化を行う例が増えているそう。オープンサイエンスの力を感じますね。
また、スライドp19「データに溺れないために:データ解析の鉄則」はMDシミュレーションの解析に限らない大事な考え方のように思いました。
紹介されていた研究の実例はこちら。薬剤耐性化に関与する多剤排出トランスポーター AcrBの機構の解明です。

- 論文:Y. Matsunaga, T. Yamane, T. Terada, K. Moritsugu, H. Fujisaki, S. Murakami, M. Ikeguchi, and A. Kidera,
“Energetics and conformational pathways of functional rotation in the multidrug transporter AcrB”,
eLife 7, e31715 (19 pages) (2018) DOI: 10.7554/eLife.31715- 日本語解説:「全原子分子動力学シミュレーションが解き明かす多剤排出トランスポーターAcrBの薬剤排出機構」 生物物理 2019(59)084-087
(小さな)プロトン化状態の変化で(大きな)膜貫通部位の運動がおこるというのは驚きです。タンパク質は本当によくできていますね。
2. この記事で試す解析とパッケージ
タンパク質の動的な挙動の理解はとても魅力的ですが、とても素人に手を出せるものではないので、ここでは基本的な解析手法の動かし方を試すことにします。
具体的には以前このブログでも取り上げたMaking-it-rainを参考に以下の解析を行います。

また利用するMDトラジェクトリ解析パッケージはMDAnalysisです。

MDAnalysisはTeachOpenCADDのT20・Analyzing molecular dynamics simulationsでも使われていて、Pythonから利用できます。また、チュートリアルが充実かつ開発コミュニティが活発そうな感じなのも魅力的です。あとロゴが可愛い。
インストールはpipかcondaでOKです(MDAnalysis - Installation)。
mamba install mdanalysis -c conda-forge
また、トラジェクトリの可視化を行うためにNGLviewを利用します(GitHub, Document)。
こちらもpipかcondaでインストールできます。
mamba install nglview -c conda-forge
mamba速い。
3. シミュレーション全体の様子を眺めよう
3-1. NGLviewerで動画を見よう
冒頭に挙げた松永先生のコロキウムでは、まず最初に「シミュレーションがうまくいったか?動画で眺めること。」を推奨されていました。失敗しているとタンパク質が崩壊するそうです。
動画で確認するために、MDAnalysisを利用してシミュレーション結果のファイルを読み込みましょう。前回の記事(Part 4)で得られたファイルは、「① OpenMMのReporterを使って出力したトラジェクトリのDCDファイル」と「②PDBファイル形式の前処理後の系の構造」です。
MDAnalysisはUniverseと呼ばれるオブジェクトを中心のデータ構造としてさまざまな処理を行う構成になっているようです(MDAnalysis tutorial - Basics)。
Universeオブジェクトはtopologyとtrajectoryを読み込んで構築できます。
import MDAnalysis as mda # topologyとtrajectoryからUniverseオブジェクトを構築 md_universe = mda.Universe("simulation_data/3poz-processed.pdb", "simulation_data/trajectory.dcd") print(type(md_universe)) # <class 'MDAnalysis.core.universe.Universe'>
次に、Jupyter notebook上で動画を確認するためのツールとしてNGL viewerのnglviewを利用します。
nglviewにはさまざまなファイルを可視化する機能があらかじめ用意されており、show_ファイル形式みたいな感じで使えます(nglview : Usage)。
MDAnalysysを使うならshow_mdanalysys()として、Universeオブジェクトを渡せばOKです。
import nglview as nv view = nv.show_mdanalysis(md_universe) view

ぱっと見、大きな崩壊はなさそうですね!よかった。
自動で水やイオンを消して見やすくしてくれるので便利です。
3-2. NGLviewが表示されない?
もしviewを実行してもjupyter notebookに何も表示されない場合は以下をお試しください。
jupyter notebookのバージョンがv5.0の時は原子数の多い大きな系を入力できなくなっている可能性があるので、ノートブック立ち上げ時にフラグを追加して上限を増やしてあげます。
jupyter notebook --NotebookApp.iopub_data_rate_limit=10000000
もしくはwidgetnbextensionやnglviewをノートブックで使えるようにアクティベートする必要があるかもしれません。
NGLviewのGitHub READMEには以下のコマンドが書かれています。*2 *3*4
python -m ipykernel install --sys-prefix jupyter nbextension enable --py --sys-prefix widgetsnbextension jupyter nbextension enable --py --sys-prefix nglview
私の環境では以上を実行すると表示されるようになりました。
3-3. エネルギー変化のプロット
今回のシミュレーションではポテンシャルエネルギー(kJ/mol)や温度(K)の継時的な変化を「log.txt」ファイルに書き出しました。
具体的な解析の前にこちらも確認しておきましょう。タブ区切りなのでPandasのread_table()でOKです。
import pandas as pd log_df = pd.read_table("simulation_data/log.txt") log_df.head()

import matplotlib.pyplot as plt x = log_df["Step"] y = log_df["Potential Energy (kJ/mole)"] plt.plot(x, y) plt.title("change of the potential energy") plt.xlabel("steps (per 0.002ps)") plt.ylabel("potential energy [kJ/mol]") plt.show()

横軸がStepで縦軸が系のポテンシャルエネルギーです。一定の範囲内で揺れ動いているようです。
今回のシミュレーションではStep Sizeを「0.002 ps」として、「500,000 steps」計算しているので1 ナノ秒のシミュレーションです。1000 steps毎にログを出力しているのでデータポイントは2 ps毎の500個になります。
温度が保たれているかプロットしておきましょう。
x = log_df["Step"] y2 = log_df["Temperature (K)"] plt.plot(x, y2) plt.title("change of the temperature") plt.xlabel("steps (per 0.002ps)") plt.ylabel("temperature [K]") plt.show()
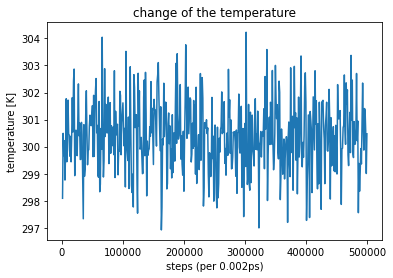
設定の300Kから-4K ~ +4K程度の範囲内に収まっていました。これは十分に温度一定のアンサンブルとみなして良いのでしょうか??
よくわからないので有識者の方ご教示いただけると嬉しいです。
4. MDAnalysisでトラジェクトリ解析
なんとなく上手くいってそう(平衡状態のサンプリングができていそう)なので、MDAnalysisを使ったより具体的な解析に進みます!
4-1. MDAnalysisのオブジェクトと解析対象の選択
まず最初にMDAnaysisにおいて一番基本的なオブジェクトについて触れておきます。
先に述べた通りMDAnalysisでは「topologyとtrajectoryから構築するUniverseオブジェクト」に対してさまざまな操作を行うことで解析を行います。
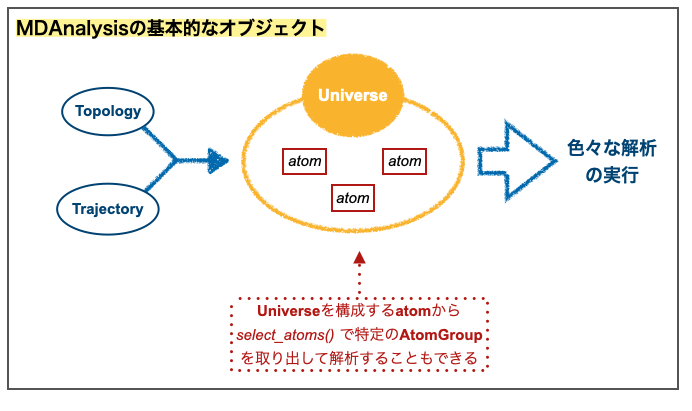
Universeの重要な属性の一つがUniverse.atomsです。これはMDAnalysisの「最も基本的な構成要素であるAtomオブジェクト」のリスト(AtomGroup)です。読んで字のごとく系を構成する原子ですね。
例えば「①Universeオブジェクト、② atomsの最初の一つ、③ atomsの最後の一つ」を取り出してみるとこんな感じです。
print("whole system : ", md_universe) print("example atoms : ", md_universe.atoms[0]) print("example atoms : ", md_universe.atoms[-1]) # whole system : <Universe with 57648 atoms> # example atoms : <Atom 1: N of type N of resname GLN, resid 701 and segid A and altLoc > # example atoms : <Atom 57648: Cl of type Cl of resname CL, resid 20103 and segid 4 and altLoc >
「① 系全部で57648原子」あり、その中には「②タンパク質の701番残基GLNの窒素原子(N)」や「③加えたイオンの塩素原子(CL) 」があることがわかります。
MDシミュレーションのトラジェクトリ解析では「系のうち特定の要素(タンパク質の特定の残基、原子、リガンド)に焦点を当てて時間変化を追跡したい!」といったことが多いようです。
このような時にはMDAnalysis.Universe.select_atoms()メソッドを使うことで、引数で指定した条件のAtomGroupを選択して解析を行うことができます。
例えば「① タンパク質 ("protein")、② 主鎖("backbone")、③ Cα原子 ("name CA")」を選択したい場合は以下のようになります。
print("protein atoms : ", len(md_universe.select_atoms("protein"))) print("backbone atoms : ", len(md_universe.select_atoms("backbone"))) print("CA atoms : ", len(md_universe.select_atoms("name CA"))) # protein atoms : 5120 # backbone atoms : 1268 # CA atoms : 317
それぞれ選択した後lenでatomの数を数えています。「②主鎖の原子数」が「③Cα原子」のちょうど4倍になっているので正しく選択できていそうです。

今回対象としているタンパク質(EGFR)のデータは「PDB ID : 3POZ」です。上図の通り前処理の段階でN末端とC末端を削っているので、シミュレーション構造には317残基残っています。Cα原子の数に一致しているようで良かった!
select_atoms()の引数には他にも色々な選択肢があるようです。ドキュメントの3. Selection commandsなどをご参照ください。
4-2. アラインメント
MDAnalysisの基本的な構成がわかったところで、解析の準備段階として、トラジェクトリにおけるタンパク質のアラインメントをとります。
TeacOpenCADD T20によると、(今回のように)シミュレーション時間が短い時はあまりおこらないそうですが、タンパク質が回転したり移動したりすることがあるそうです。大きく位置がずれると解析が難しくなるのであらかじめアラインメントをとって揃えておく方が良いそうです。
また、MDAnalysisの「ドキュメント: Alignments and RMSfitting」にも、「RMSDやRMSFといった値が意味のあるものにするためにはトラジェクトリのアラインメンとを取ること」との記述があります。
アラインメントにはMDAnalysis.align.AlignTrajを使います(Aligning a trajectory to a reference)。ここではトラジェクトリの最初の構造をリファレンスとして全体を合わせます。
from MDAnalysis.analysis import align # トラジェクトリの位置を最初のフレームにリセットする md_universe.trajectory[0] # align.AlignTrajでアラインメントを取る alignment = align.AlignTraj( mobile = md_universe, reference = md_universe, select="protein", in_memory=True) alignment.run()
動画で見ます。
view = nv.show_mdanalysis(md_universe) view

・・・違いがわからない。
AlingTrajの引数ではmobileで動かす対象のUniverseオブジェクトを指定し、referenceのUniverseオブジェクトに合わせます。上記ではどちらも同じUniverseオブジェクトを指定してますが、実行前にトラジェクトリの最初のフレームにリセットする操作を行なっているため、開始時点の位置に対して全体のアラインメントをとることになっています。
また、引数selectではトラジェクトリのうち「どの構造のアラインメントを取るか」選択することができます。上記では「"protein"」とすることでタンパク質全体を選んでいます。先に見たように「"name CA"」でCA原子(アミノ酸Cα炭素原子)を選択することもできます。
最後に、今回は引数「in_memory=True」とすることでその場でアラインメントをとったものに置き換えていますが、トラジェクトリのサイズが大きくてメモリに載せきれない場合は、一度ファイルに書き出して再度トラジェクトリを読み込むといった処理もできます。
以下のようにすればOKです。
# 検証用にuniverseオブジェクトを作成 md_universe2 = mda.Universe("simulation_data/3poz-processed.pdb", "simulation_data/trajectory.dcd") md_universe2.trajectory[0] # アラインメントをファイルに書き出す場合。 alignment2 = align.AlignTraj(mobile = md_universe2, reference = md_universe2, select='name CA', filename='simulation_data/CA_aligned.dcd', in_memory=False) alignment2.run()
CA原子でアラインメントをとって「CA_alinged.dcd」ファイルに書き出しました。*5読み込んで可視化します。
md_universe3 = mda.Universe("simulation_data/3poz-processed.pdb", "simulation_data/CA_aligned.dcd") view = nv.show_mdanalysis(md_universe3) view

動画のどこが違うがお分かりでしょうか?・・・私にはさっぱりわかりません。
4-3. RMSD
アラインメントも無事とれたので、とっつきやすいRMSD(wikipedia - 原子位置の平均二乗偏差)から解析を行なってみましょう!
だいたいこういう話です。*6

少し注意点ですが、MDAnalysisのメソッドには小文字のrmsdと大文字のRMSDがあり、解析機能(や引数)が異なります。
4-3-1. フレーム2点間のRMSD(rms.rmsd())
まず、RMSDの評価としてトラジェクトリの「フレームの最初と最後でどれだけ構造変化があったか?」を検証してみます。2つの状態の比較の場合はMDAnalysis.analysis.rms.rmsd()を使えば良いようです。rmsdが小文字であることにご注意ください(MDAnalysis Tutorial : 8. Using the MDAnalysis.analysis modules)。
from MDAnalysis.analysis import rms # 最初のフレームにリセット md_universe.trajectory[0] # 最初のタンパク質の位置 protein_first = md_universe.select_atoms("protein").positions # 最後のフレームに移動 md_universe.trajectory[-1] # 最後のタンパク質の位置 protein_last = md_universe.select_atoms("protein").positions # rmsdを評価 print(rms.rmsd(protein_first, protein_last)) # 1.7643614273332058
1.76Å・・・なるほど?・・・この値がどの程度なのか良くわかりません。
4-3-2. トラジェクトリ全体のRMSD推移(rms.RMSD())
次にタンパク質の主鎖(backbone)に焦点を当てて、トラジェクトリ全体での変化を見てみましょう。全体を見る場合はMDAnalysis.analysis.rms.RMSD()を使います(大文字のRMSDに注意!)。
# フレームのリセット md_universe.trajectory[0] # RMSD解析 RMSD_analysis = rms.RMSD(md_universe, # アラインメントを取るオブジェクト md_universe, # アラインメントを合わせるリファレンスのオブジェクト select = "backbone", # 計算対象を主鎖にする ref_frame = 0 # リファレンスのフレームインデックス ) RMSD_analysis.run()
run()で解析を実行すると、結果はresults.rmsdに「[[frame, time(ps), RMSD(A)], [...], [...]]」という情報を含むnumpy.ndarrayの形で保存されています。
print("type : ", type(RMSD_analysis.results.rmsd)) print("1st data : ", RMSD_analysis.results.rmsd[0]) print("total data number : ", len(RMSD_analysis.results.rmsd)) # type : <class 'numpy.ndarray'> # 1st data : [0.0000000e+00 0.0000000e+00 6.0600393e-07] # total data number : 50
matplotlibでプロットしましょう。0 psはリファレンスそのものなので除きます。
time_ps = RMSD_analysis.results.rmsd[1:, 1] backbone_RMSDs = RMSD_analysis.results.rmsd[1:, 2] plt.plot(time_ps, backbone_RMSDs) plt.title("change of the RMSD") plt.xlabel("Time (ps)") plt.ylabel("RMSD [$\AA$]") plt.show()
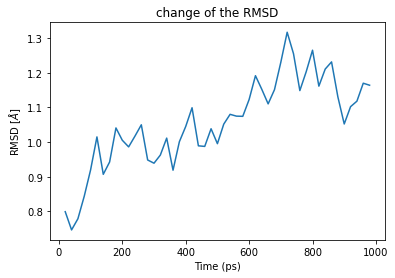
右肩上がりのプロットになりました。600 ps以降は1.1Å前後をうろうろしている感じです。
ついでにRMSDの分布もプロットしてみましょう。seabornのkdeplotを使ってカーネル密度推定法のプロットを行います。
import seaborn as sb sb.kdeplot(RMSD_analysis.results.rmsd[1:, 2], shade=True) plt.title("distribution of the RMSD") plt.xlabel("RMSD [$\AA$]") plt.show()

トラジェクトリ初期のリファレンスとのRMSDが小さい状態は短く、後半のRMSDが1.1Å前後まで広がった状態が多くを占める様子がわかりました。
4-4. RMSF
4-4-1. RMSFって?
つづいてRMSF(平均二乗揺らぎ, Root-mean-square fluctuation)を計算します。これは分子内原子の平均位置からの分散を示す量で、平衡状態からのトラジェクトリ構造の揺らぎを表すそうです。*7

4-4-2. RMSFの計算
RMSFの計算にはMDAnalysis.analysis.rms.RMSF()を使います。引数には計算したいatomgroupを指定します。
アラインメントが取られたタンパク質を使うことが想定された機能なので先のalign.AlignTrajを実行していない場合は処理を忘れずに!
Cα原子についてRMSFを計算する場合はこんな感じです。
# Cαの選択 C_alphas = md_universe.select_atoms("name CA") # RMSF計算 RMSF_analysis = rms.RMSF(C_alphas) RMSF_analysis.run()
run()で解析を実行すると、結果はresults.rmsfにnumpy.ndarrayとして格納されます。RMSDのときと同じですね。
結果をプロットしましょう。Cα原子の残基番号はresnumsで取り出せます。
# 残基番号の取り出し(resnums) residue_numbers = C_alphas.resnums # プロット plt.plot(residue_numbers, RMSF_analysis.results.rmsf) plt.title("C_alpha RMSF") plt.xlabel("residue number") plt.show()
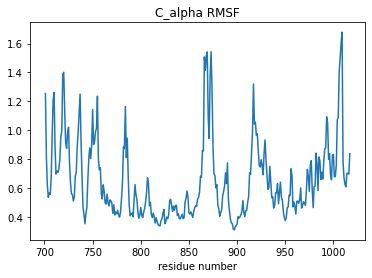
両末端でRMSF(揺らぎ)が大きくなっているのは納得として、850番 ~ 900番にも揺らぎのピークがあります。具体的にはEGFRタンパクのどのあたりの構造なのでしょうか?面白そうなのでもう少し調べます。
4-4-3. 文献計算例との比較
今回のシミュレーションのRMSF解析の結果が妥当か?文献計算例と比較してみましょう。
まず、具体的なタンパク質構造に戻って確認してみます。PyMolで該当の領域(850 ~ 900)を色分けしてみました。
だいたいこの辺。

キナーゼ構造でいうところのActivation loop(A-loop)に相当しそうです。
(ググって最初に出てきた文献と)結果を比較してみましょう。
Tamirat MZ, Koivu M, Elenius K, Johnson MS
Structural characterization of EGFR exon 19 deletion mutation using molecular dynamics simulation.
PLoS ONE 2019(14): e0222814
EGFRの構造を漫画で描くと以下で、DFGモチーフ(D855, F856, G857)に続いてA-loopと呼ばれる構造があるそうです。

上記論文は野生型(Wild EGFR)と変異型(欠損の変異、Δ746ELREA750)についてMDシミュレーションを行なって、変異が構造に与える影響を調べたもののようです。詳細は論文をご参照いただくとして、RMSFの評価を引用します。

野生型、変異型ともにA-loopでRMSFが大きくなっています!また、値としても1〜 2Åということで近い値でした。
素人目には類似の結果が得られているようにみえるのですが、専門家の目からみるとどうなんでしょうか?ご教示いただけると嬉しいです。
4-5. 回転半径(radius of gyration)
最後に回転半径の計算を行ってみます。こういう感じ指標です。*8

回転半径の算出にはAtomGroupに紐づいたメソッドであるradius_of_gyration()を使えば良さそうです。例えばCα原子の回転半径を求めるならこんな感じ。
# フレームのリセット md_universe.trajectory[0] # Cαの選択 C_alphas = md_universe.select_atoms("name CA") # 回転半径の計算 C_alphas.radius_of_gyration() # 19.713873743719788
19.7Åと出ました。select_atoms()で目的の原子群を選択してAtomGroupを作っておいてメソッドを適用するのはこれまで何度か出てきた通りですね。
トラジェクトリ全体を通した変化を見たい場合は、Universeオブジェクトのtrajectory属性についてforループを回してフレームをイテレーションするのが簡単です。*9*10
# フレームを格納するリスト frame_list= [] # 回転半径を格納するリスト rgyr_list = [] # トラジェクトリのフレームごとに回転半径を計算してリストに格納 for ts in md_universe.trajectory: f = ts.frame rgyr = C_alphas.radius_of_gyration() frame_list.append(f) rgyr_list.append(rgyr) # プロット plt.plot(frame_list, rgyr_list) plt.title("change of the radius of gyration") plt.xlabel("frame") plt.ylabel("rgyr [$\AA$]") plt.show()

「1 ns ( = 0.002(ps) x 500,000 (steps))」のシミュレーションが計50フレームなので「1フレーム = 20 (ps) = 10,000 (steps)」です。上下はあるものの途中までは継時的に増加し、19.8Å付近で平衡にあるように見えます。RMSDで見た挙動に似ていますね。
回転半径の分布もプロットしてみましょう。
sb.kdeplot(rgyr_list, shade=True) plt.title("distribution of the radius of gyration") plt.xlabel("rgyr [$\AA$]") plt.show()
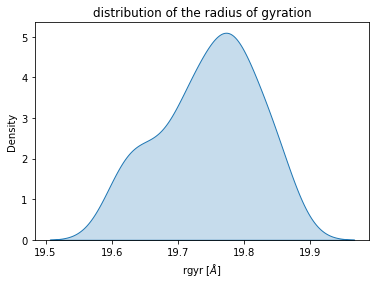
回転半径の分布はRMSDの分布と比較して分布の肩の部分が高くなっています。同じトラジェクトリでも指標によってばらつきの度合いが変わるのは面白いですね。(・・・生物学的な解釈はわかりませんが、、、)
尚、ドキュメント(MDAnalysis User Guide:Radius of gyration)には回転半径を求める関数を自作する例が書かれています。ご興味のある方はご参照ください。
5. おわりに
以上、今回の記事ではMDシミュレーショントラジェクトリの解析を試みました。
冒頭に挙げたコロキウム資料を参考に、①Jupyter notebook上でNGLViewを使った動画の確認を行ったのち、②MDAnalysisを使ったRMSD、③RMSF、④回転半径の算出を行いました。
解析操作で忘れがちな注意点としてはアラインメントをとることとトラジェクトリのフレーム位置のリセットでしょうか?
何はともあれ、素人でもチュートリアルで動かせるOpenMMやMDAnalysisはとても素晴らしいですね!オープンソースコミュニティに感謝です!
個人的にはRMSFでEGFRキナーゼのA-loopの揺らぎが確認できた点が非常に面白かったです。また、RMSDと回転半径で分布が変わってくる点も興味深かったです。
今回取り上げたもの以外にもMDトラジェクトリの解析には色々とあるようです。続きはCM②のあとで!
よく理解せずにまま実行動かしているので間違いが多いと思います。ご指摘いただければ幸いです。
ではでは
*1:下図のMDAnalysisのロゴはドキュメントから。イラストはいらすとやさんより。
*2:NGLviewのDevelopment versionのインストール方法の部分です。
*3: フラッグ--sys-prefixは現在の(アクティベートされてる)仮想環境にkernelをインストールするためのもののようです。
*4:参考:How to properly use --sys-prefix。kernelのインストールがnglviewのアクティベートに必須かはよくわかりませんでした。
*5:引数「in_memory=True」はmobileで指定したトラジェクトリを永久にメモリ上のトラジェクトリ(in-memory trajectory)に変えるもののようです。そのためか、一度align.AlignTrajを実行したUniverseオブジェクトに再度実行してもファイルの書き出しが行われませんでした。そこでここでは再度オブジェクトを作り直しています。
*6:図の記載は理化学研究所 計算科学研究センターのeラーニング「計算生命科学の基礎6 生体系分子シミュレーションの新展開」における横浜市立大学 池口満徳先生(研究室HP)の講義動画(YouTube)とスライド資料(SlideShare)などを参考にしています
*7:こちらも図の記載は先のeラーニング資料などを参考にしています。
*8:こちらも図の記載は先のeラーニング資料などを参考にしています。
*9:参考:MDAnalysis:Overview over MDAnalysis
*10:フレームのイテレーションについてはMDAnalysis UserGuide:Slicing trajectoriesなども参考になります。