molzipで色々なリンカーのPROTACをつくろう!
RDKitのとても便利な機能molzipというのを知りました。複数のフラグメントをまとめてひとつ分子を構築できるというもので、① @dr_greg_landrum先生の記事(R-group decomposition and molzip)と、② @iwatobipen先生の記事(Build peptide from monomer library from ChEMBL)に使い方が詳しく紹介されています。
①の記事ではRグループ分析と組み合わせた分子ライブラリの構築方法を、②の記事ではモノマーを組み合わせたペプチドライブラリの構築方法を紹介してくださっています。
ユニットを組み合わせて作るタイプの分子に威力を発揮しそうですね!ユニットといえば他に思いつくのは・・・PROTAC!!
というわけで、molzipを使って色々なリンカーのPROTACを作ってみたいと思います!また、構造データの取得元としてPROTAC-DBを参照します。

※ 注)この記事でやることは基本的に上記2記事と一緒です。遊んでみたかっただけです。手抜きでごめんなさい。
1. molzipの基本
1-1. 基本的な使い方-1
まずはmolzipの基本的な使い方を確認しましょう!molzipのドキュメントはこちら(RDKit documentation :molzip)。
こんな感じで別々の分子(Molオブジェクト)を1つにまとめる関数です。

「どこで分子どうしをつなぐか?」はオプションの引数(MolZipParams)で設定されます。デフォルトではMolzipLabel.AtomMapNumberとなっています。
あらかじめそれぞれの分子の結合を作りたい原子に目印(ダミーアトム + ナンバリング)をつけておいて、それらの原子同士をつなぐ(ジッパーでとめる)感じです。
・・・チャックするってもう言わないの???
1-2. molzipの使用例-1
簡単な使用例を見てみましょう!Buchwald-Hartwigクロスカップリングをしたいと思います。

C-N結合をつくりたいので、上図のように反応点をダミーアトム(*)にしてナンバリングしたMolオブジェクトを作りましょう!
from rdkit import Chem mol1 = Chem.MolFromSmiles('c1ccccc1[*:1]') mol2 = Chem.MolFromSmiles('C1CCCCN1[*:1]') Chem.Draw.MolsToImage([mol1, mol2])

できました!ワイルドカードのアスタリスク(*)にコロン(:)を挟んで数字を置いて、角格好([ ])で囲えばOKです。*1
あとはmolzipするだけ。
product = Chem.molzip(mol1, mol2) Chem.Draw.MolToImage(product)

マッピングした原子同士で結ばれた分子ができました!
1-3. 基本的な使い方-2
さて、2つのMolオブジェクトをつなぐという使い方でした。
molzipには他にも「複数のフラグメントが1つのMolオブジェクトに入ったものを対象にして分子を作る」、といった使い方もあります。
例えば、以下のようにSMILESをピリオド(.)を挟んで繋げれば複数のフラグメントが入ったオブジェクトを作れます。
mol3 = Chem.MolFromSmiles('[*:1]c1ccccc1.C1N([*:1])CC(C)(C)N([*:2])C1.c1nc([*:2])ccc1')
Chem.Draw.MolToImage(mol3)

ベンゼンとピリジンが近接していて見づらいですが、フラグメント3つと2種類のアトムマッピング(1,2)を含んでいます。
これをmolzipするとこんな感じ。
product2 = Chem.molzip(mol3) Chem.Draw.MolToImage(product2)

それぞれ指定したマッピングの箇所でつながった分子ができました!3つ以上のフラグメントをつないで分子を構築したい場合に便利そうです。
2. この記事でやること
molzipの使い方がわかったところで、この記事の目標です。
タンパク質分解誘導キメラ分子(Proteolysis targeting chimeda、PROTAC)は、① 分解したい対象のタンパク質に結合するWarheadと、② ユビキチンリガーゼに結合するE3 ligand、そしてをつなぐLinker(③)の3つの要素から構成されています。

「何を分解するか?(Warhead)」「どうやって分解するか?(E3 ligand)」という点に注目がいきがちですが、細胞内で分解機能を発揮するためには適切なリンカーを見つけ出すことが重要だそうです。
上図に引用した下記文献では、それぞれの標的ごとに試行錯誤しなければいけないリンカーについて焦点を当てて議論されています。(オープンアクセス, CC BY 4.0)*2
Troup RI, Fallan C, Baud MGJ.
Current strategies for the design of PROTAC linkers: a critical review.
Explor Target Antitumor Ther. 2020;1:273-312.
例えばこんな構造があるそう。

さらに組み合わせたり、長さが違ったり、色々あって大変だ!・・・それなら、どんな組み合わせ構造があるか全部作っちゃえばよくない?
というわけで、いろんなリンカーと残りのユニット(Warhead、E3 ligand)の組み合わせをmolzipでつくってみましょう!
3. PROTAC DBから構造を取得しよう
3-1. PROTAC DBって?
さて、いろいろな組み合わせ構造をつくろう!といっても、元になる構造群がないとどうにもなりません。
「どこかにまとまっていないかな?」・・・ということで素敵なデータベースがありました。その名もPROTAC-DB!!
文献は以下(オープンアクセス, CC BY 4.0)
Gaoqi Weng, Chao Shen, Dongsheng Cao, Junbo Gao, Xiaowu Dong, Qiaojun He, Bo Yang, Dan Li, Jian Wu, Tingjun Hou,
PROTAC-DB: an online database of PROTACs,
Nucleic Acids Research, Volume 49, Issue D1, 8 January 2021, Pages D1381–D1387
Fig. 1を引用します。

色々な文献からPROTACの例をあつめてきて活性データといっしょにまとめたデータベースのようです(PROTACにフォーカスしたChEMBLみたいな感じ?)。
さらに素晴らしいことに、それぞれのPROTACをWarhead、Linker、E3 Ligandのユニットに分割した構造のアノテーションもしてくださっているようです。これは便利!
3-2. PROTAC DBにアクセス!
早速アクセスしてみましょう。URLはこちら(「PROTAC-DB : http://cadd.zju.edu.cn/protacdb/ 」)
こんな感じでした。

最新版のリリースは2021-04-17で、約2000個の分子が登録されています。
例としてArvinas社のBET degrader、ARV-771を検索してみましょう*3。結果のサマリーはこんな感じ。

標的タンパク(BRD2, BRD3, BRD4)やE3ユビキチンリガーゼ(VHL)、種々の表記(IUPAC、InChI etc.)といった情報があります。また、構造を3つのユニットに分けて分類していることも確認できました。
スクロールダウンすると、RDKitで計算したプロパティ(MW, logP, TPSA etc.)や、活性値といった情報も見ることができます。
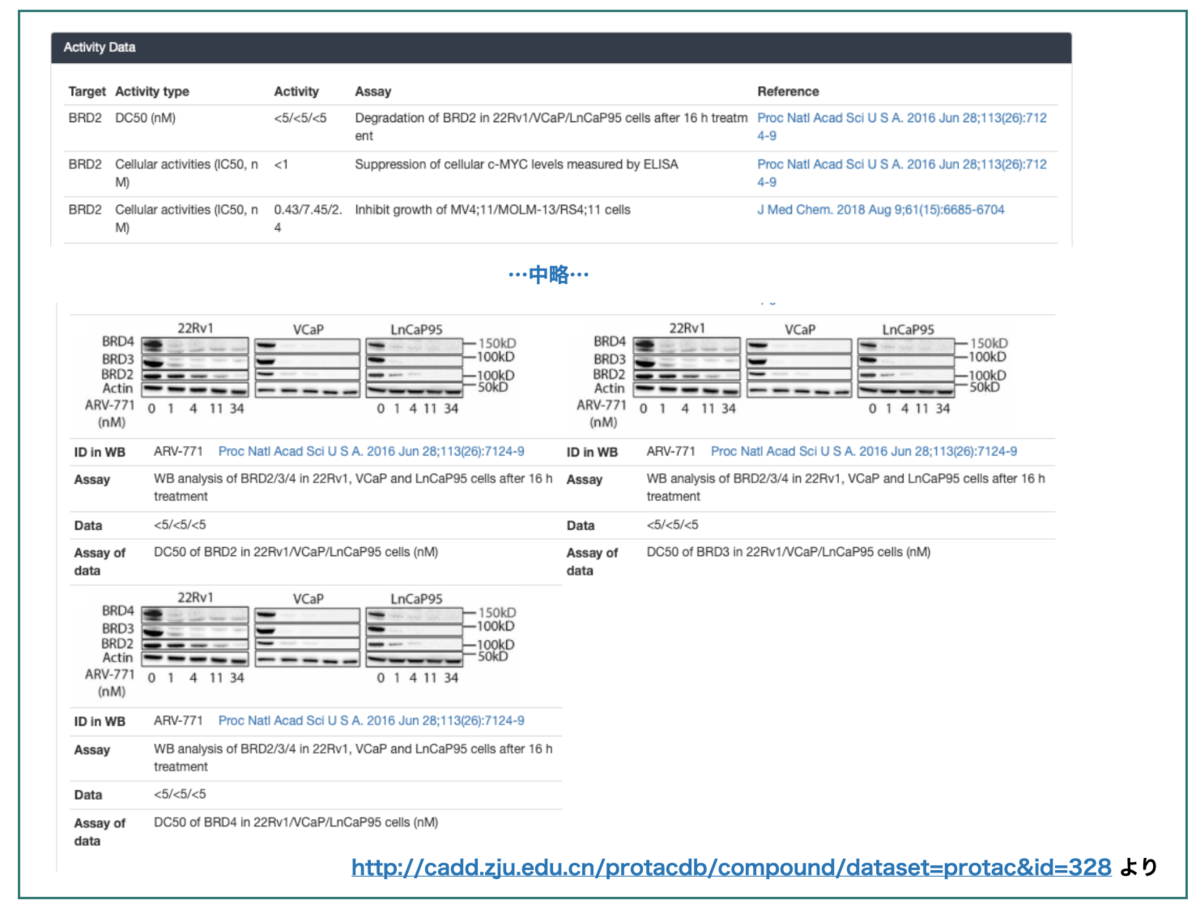
ウェスタンブロットの図まであるのにびっくり!元の文献にもReferenceをクリックで飛べます。素敵!
3-3. データをダウンロード
それでは目的の構造情報を取得しましょう!
下図のようにDownloadsをクリックして、移行先のページでDATA SOURCESから目的のユニットを選びます(今回はLinkers)。

表示されるテーブルは標的タンパクごと(Compoud Group)にまとめられていて、SDF形式あるいはCSV形式でダウンロードすることができます。
今回はDB上にある全てのリンカーの情報が欲しいので「All行のlinker.csv」をダウンロードしました。
ダウンロードしたCSVファイルを確認してみます。

上図のように構造(SMILES)、その他プロパティ計算値が格納されています。
SDFではなくCSVを選んだポイントは「Smiles_R列」です。結合箇所が「[R1]、[R2]」で示されていて分かりやすくなっています。SDFではこの情報がないため、「どこを結合箇所にすればよいか?」わからなくなっています。
先に見た通りmolzipの処理では、結合箇所の指定(AtomMapNumber)が大事でした。ですのでCSVファイルの「Smiles_R」のように結合箇所のわかりやすい情報の方が使いやすそうです。
何はともあれ、これでリンカーの情報が手に入りました。
4. molzipでいろいろなリンカーのPROTACをつくろう
4-1. 基本骨格の選択(ARV-771)
それでは色々なリンカーのPROTACをつくってみましょう!
まず、今回はLinker以外の①Warhead、②E3 ligandは固定したいと思います。
先にPROTAC-DBで見たARV-771を基本骨格として利用することにしましょう。
Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, Wang J, Chen X, Dong H, Siu K, Winkler JD, Crew AP, Crews CM, Coleman KG.
PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer.
Proc Natl Acad Sci U S A. 2016 Jun 28;113(26):7124-9.
PROTAC-DBの該当ページ(ID:328)からcanonical SMILESをコピペしてRDKitに読み込ませます。
ARV771 = Chem.MolFromSmiles('CC1=C(C2=CC=C([C@H](C)NC(=O)[C@@H]3C[C@@H](O)CN3C(=O)[C@@H](NC(=O)COCCCOCCNC(=O)C[C@@H]3N=C(C4=CC=C(Cl)C=C4)C4=C(SC(C)=C4C)N4C(C)=NN=C34)C(C)(C)C)C=C2)SC=N1')
Chem.Draw.MolToImage(ARV771)

4-2. R-group分析で3つのユニットに分解
これを3つのユニットに分けましょう。リンカーをコア構造にしてR-group decompostionすれば良さそうです。参考のRDKit blog記事の通りやればOK!
まずコア構造をPROTAC-DBのARV-771 Linker構造をもとに作ります。
ARV771_linker = Chem.MolFromSmiles('NCCOCCCOC')
Chem.Draw.MolToImage(ARV771_linker)

R-group decompositionします(参考:RDKit ドキュメント rdRGroupDecompose)。
from rdkit.Chem import rdRGroupDecomposition rgroups, _ = rdRGroupDecomposition.RGroupDecompose([ARV771_linker], [ARV771], asRows=False)
引数は①コア構造、②対象の分子の順番で、それぞれ複数の構造を入力できるようになっているので、リストにして渡しています。
結果は辞書型なので、keyを確認しましょう。
print(rgroups.keys()) # 出力 # dict_keys(['Core', 'R1', 'R2'])
Core、R1、R2とラベル付けされた3つに分解されました。
辞書のvalueはMolオブジェクトを格納したリストです。描画します。
Chem.Draw.MolsToGridImage([rgroups['Core'][0], rgroups['R1'][0], rgroups['R2'][0]], legends=['Core', 'R1', 'R2'])

うまくできていそうです。
4-3. 結合部位の再ラベル化
このままリンカーとの連結に進みたいところですが、WarheadがR2、E3 ligandがR1となっています。PROTAC-DBと結合点のナンバリングがずれてしまうので、「Warhead : 1」「E3 ligand : 2」となるように修正しましょう。
WarheadのMolオブジェクトを確認すると、結合点のAtomのSymbolはR2になっています。
ARV771_warhead = rgroups['R2'][0] for atom in ARV771_warhead.GetAtoms(): if atom.GetSymbol() == "R2": for prop in atom.GetPropNames(): print(prop, ':', atom.GetProp(prop)) # 出力 # molAtomMapNumber : 2 # dummyLabel : R2
上のように、プロパティとして「dummyLabel : R2」「molAtomMapNumber : 2」が付与されていることがわかります。SetPropを使ってラベルを「* : any atom」、ナンバリングを「1」にしましょう。
for atom in ARV771_warhead.GetAtoms(): if atom.GetSymbol() == "R2": atom.SetProp('dummyLabel', '*') atom.SetProp('molAtomMapNumber', '1') Chem.Draw.MolToImage(ARV771_warhead)

できました!
同様にして、E3 ligandについてR1を「dummyLabel : *」「molAtomMapNumber : 2」に変更します。
ARV771_E3 = rgroups['R1'][0] for atom in ARV771_E3.GetAtoms(): if atom.GetSymbol() == "R1": atom.SetProp('dummyLabel', '*') atom.SetProp('molAtomMapNumber', '2') Chem.Draw.MolToImage(ARV771_E3)

見づらいですが、右端が「* : 2」になっています。
4-4. リンカーライブラリの作成
WarheadとE3 ligandが準備できたので色々なリンカー部位のデータベースを作成します。
PROTAC-DBからダウンロードした「linker.csv」をPandasのDataFrameに読み込みます。
import pandas as pd linker_df = pd.read_csv('linker.csv')[['Compound ID', 'Smiles_R']]

先に見た通り「Smiles_R列」に結合箇所を「[R1]、[R2]」で明記したSMILESが格納されています。これを「[* : 1]、[* : 2]」に置き換えましょう。
テキストを置換する関数をつくってmap()でDataFrameに適用します。
def linker_converter(smilesR): r1 = smilesR.replace('[R1]', '[*:1]') r2 = r1.replace('[R2]', '[*:2]') return r2 linker_df['smiles_dummy'] = linker_df['Smiles_R'].map(linker_converter)

ダミーアトムに置換したSMILESが作成できていそうです。これでリンカーライブラリができました。
4-5. ユニットを連結しよう!
だいぶんと長くなってしまいましたが、やっと3つのユニットの基礎となる情報が揃いました。あとはひたすら繋いでいくだけです。
molzipの基本的な使い方で見た通り、3つのユニットをつなぐにはフラグメントをまとめて1つのMolオブジェクトにしておけばOKでした。
参考記事(R-group decomposition and molzip)では、RWMolオブジェクトに変更した後、Insert()してフラグメントをまとめていますが、ここでは簡単のためSMILESを連結する方式にします。
WarheadとE3 ligandをそれぞれSMILESに変換して、linkerのSMILESと「ピリオド(.)」を挟んでつないでいきます。
warhead_smiles = Chem.MolToSmiles(ARV771_warhead) E3_smiles = Chem.MolToSmiles(ARV771_E3) def linker_connector(linker_smiles): connected_smiles = warhead_smiles + '.' + linker_smiles + '.' + E3_smiles return connected_smiles linker_df['protac_smiles'] = linker_df['smiles_dummy'].map(linker_connector) print(len(linker_df)) # 出力 # 1099

SMILESが長すぎて出来ているのかよくわからない。。。とりあえず1099行あるようです。
あとは1つずつ取り出してMolオブジェクトに変換した後、molzipしていきます。
molzipに失敗する場合もあるので、成功したものを「リスト:PROTACs」、失敗した場合のリンカーを「リスト:failed_linker」に格納していきます。
PROTACs = [] failed_linker = [] idx = 0 for smi in linker_df['protac_smiles']: fragments = Chem.MolFromSmiles(smi) try: protac = Chem.molzip(fragments) PROTACs.append(protac) except: fail_lnk = linker_df['smiles_dummy'].iat[idx] fail_lnk_m = Chem.MolFromSmiles(fail_lnk) failed_linker.append(fail_lnk_m) idx += 1 print('Generated PROTACs : ', len(PROTACs)) print('failed linkers : ', len(failed_linker)) # 出力 # Generated PROTACs : 1092 # failed linkers : 7
1099個のリンカーのうち、1092個でユニットを繋ぐことができ、7個で失敗しました。
4-6. できたPROTACを確認してみよう!
それではどんなPROTACができたのか確認してみましょう。
最初の3つを取り出してみます。ちょっと描画を頑張ります。*4
from rdkit.Chem.Draw import rdMolDraw2D from IPython.display import SVG view = rdMolDraw2D.MolDraw2DSVG(990,350, 330, 350) view.DrawMolecules([rdMolDraw2D.PrepareMolForDrawing(m) for m in PROTACs[:3]]) view.FinishDrawing() svg = view.GetDrawingText() SVG(svg)

鎖(-CH2CH2O-)の長さの異なるリンカーや、アミド結合の数が異なるものが出来ているようです。
もともとのARV-771のWarheadとリンカーの結合はアミド結合でした。ですので、リンカーライブラリの中で結合末端が炭素原子の場合は、Warheadとの結合がケトンになっています。あまり好ましくはないかもしれませんが、お試しということで。。。
「リンカーで多様性が出ているのか?」、記述子を計算して眺めてみましょう。
分子量(MolWt)、LogP(MolLogP)、水素結合アクセプター数(CalcNumLipinskiHBA)、水素結合ドナー数(CalcNumLipinskiHBD)を計算してデータフレームに格納します。*5
from rdkit.Chem import Descriptors, rdMolDescriptors df_desc = pd.DataFrame(columns=['MW', 'LogP', 'HBD', 'HBA']) df_desc['MW'] = [Chem.Descriptors.MolWt(m) for m in PROTACs] df_desc['LogP'] = [Chem.Descriptors.MolLogP(m) for m in PROTACs] df_desc['HBA'] = [Chem.rdMolDescriptors.CalcNumLipinskiHBA(m) for m in PROTACs] df_desc['HBD'] = [Chem.rdMolDescriptors.CalcNumLipinskiHBD(m) for m in PROTACs]
ヒストグラムをプロットします。
import seaborn as sns from matplotlib import pyplot as plt fig, axes = plt.subplots(2, 2, figsize=(10,10)) sns.histplot(df_desc['MW'], kde=True, binwidth=50, ax=axes[0,0]) sns.histplot(df_desc['LogP'], kde=True, binwidth=0.5, ax=axes[0,1]) sns.histplot(df_desc['HBA'], kde=True, binwidth=1, ax=axes[1,0]) sns.histplot(df_desc['HBD'], ax=axes[1,1])

思っていたよりも記述子にバラツキがでました。分子量では900 ~ 1500程度、LogPでも4 ~ 12前後となっています。
なお、今回もとにした化合物、ARV-771では以下の値となります。
print('MW of ARV-771 : ', Chem.Descriptors.MolWt(ARV771)) print('LogP of ARV-771 : ', Chem.Descriptors.MolLogP(ARV771)) print('HBA of ARV-771 : ', Chem.rdMolDescriptors.CalcNumLipinskiHBA(ARV771)) print('HBD of ARV-771 : ', Chem.rdMolDescriptors.CalcNumLipinskiHBD(ARV771)) # 出力 # MW of ARV-771 : 986.6619999999997 # LogP of ARV-771 : 6.531180000000008 # HBA of ARV-771 : 16 # HBD of ARV-771 : 4
リンカーライブラリと組み合わせて作ったPROTACライブラリと比較すると、一番頻度が高い範囲(より少し小さいくらい?)に含まれていそうです。
4-7. 失敗したリンカーを確認
最後に、molzipがうまくいかなかったリンカーを確認してみましょう。
view = rdMolDraw2D.MolDraw2DSVG(990, 600, 330, 200) view.DrawMolecules([rdMolDraw2D.PrepareMolForDrawing(m) for m in failed_linker]) view.FinishDrawing() svg = view.GetDrawingText() SVG(svg)

結合末端が、最初の5つは「窒素原子との2重結合( N=[* : 1])」、残りの2つは「同じAtomMapNumber([* : 2])が2つ」となっています。同じ結合箇所に2回結合を作ろうとして失敗しているようです。
エラーメッセージも「Explicit valence for atom # 18 C, 5, is greater than permitted」とでていました。「炭素の価数が5になっちゃうよ!」って怒ってます。
ごめんなさい。
5. まとめ
以上、今回はユニットで構成される化合物の例としてPROTACを題材に、RDKitのmolzip関数を使ってフラグメントを連結する方法で遊んでみました。また、PROTACで使用されるリンカー構造の取得元としてPROTAC-DBを参照しました。
生成したPROTACライブラリの記述子を計算すると、予想以上にバラツキがでました。WarheadやE3 ligandと比べるとLinkerは単純な構造に見えますが、ユニットを組み合わせてできる化合物の性質には少なからず影響を与えそうです。
今回の記事では結合形成部位の化学的性質については全く考慮しませんでした。従って、記事中で見たように元々はアミド結合だったものがケトンになっていたりとしました。結合のタイプも含めて化合物生成を行うためには、RDKitのReactionといったものを使った方が良さそうです*6。
ですが、Reaction SMILES(やSMARTS)はSMILESよりハードルが高くてしんどいので、お手軽に構造生成できるmolzipはとても便利だなーと思いました。
出てくるコードは基本的に参考にさせていただいた記事のコピペです。すみません。。。題材を変えたということで許してください。
おしまい*7
*1:このあたりおそらくSMILESではなくSMARTSの領域になると思うのですが、RDKitではMolFromSmilesで変換できます。表記方法に詳しくないのでよくわからないです。
*2:ググって一番上にでてきました。これが一般的な話かは分かりません。すみません。
*3:PROTAC-DBでARVで検索して一番上に出てきたのがこれでした
*4:参考:化学の新しいカタチさんの記事「RDKitでの構造式描画を詳しく解説」
*5:参考:化学の新しいカタチさんの記事「RDKitにおける記述子の扱い方をリピンスキーの法則を通して学ぶ」
*6:参考:化学の新しいカタチさんの記事「RDKitで化学反応:ケモインフォマティクスにおける反応式の扱い方」
*7:ところで、今回の記事の冒頭にBuchwald-Hartwigクロスカップリングを上げてみました。色々なアミンと芳香族をつなげて素晴らしい反応ですよね!アミンの種類が多ければ多いほどバラエティに富んだ構造をつくれます。 要するにビルディングブロックの多様性が、ケミカルスペースの探索空間につながるそうです。
Grygorenko, O.O., Volochnyuk, D.M., Vashchenko, B.V.
Emerging Building Blocks for Medicinal Chemistry: Recent Synthetic Advances
Eur. J. Org. Chem. 2021 6478-6510