Google CouldでMD計算をお試し!ーMaking it rainー
タンパク質がぐにゃぐにゃ動く分子動力学(MD)計算かわいいですよね。下はD.E. Shaw Research("DESRES")のツイートで、彼らの開発したスパコン(アントン - wikipedia)で計算されたものらしいです*1。
50 2-µs simulations of FDA approved or investigational drug molecules binding to a construct of the #SARSCoV2 trimeric spike protein. Full data at https://t.co/CQVDq1clZB pic.twitter.com/LEQ9RqBKLy
— D. E. Shaw Research (@DEShawResearch) 2020年5月6日
「お高いパソコンがないとMDで遊べないのかな?」と思ってましたが、Twitterで「低分子ならGoogle Colabでも結構できるよ!」「Making it rainが参考になるよ!」って教えていただきました。@ylogtさん、ありがとうございました!
というわけで、とりあえずMakig-it-rainをそのまま実行した結果のご報告です。*2

1. Making it rain?
1-1. 概要
Making-it-rainは計算リソースが必要な分子動力学(MD)計算を、誰でもお手軽にできるようにしよう!というプロジェクトです。GoogleアカウントがあればOK!
Maiking-it-rainではOpenMMというオープンソースのMDシミュレーションライブラリをGoogle Colab上で実行することができます。ノートブックに従って順番に実行することで、①MD計算の準備、②実行、③結果の解析まで行えます。また、Google Driveとの連携されているので、計算結果やノートブックの保存も容易です。
論文は以下。
- P.R. Arantes, M.D. Polêto, C. Pedebos, and R. Ligabue-Braun Making it Rain: Cloud-Based Molecular Simulations for Everyone
J. Chem. Inf. Model. 2021, 61, 10, 4852–4856
GitHubレポジトリは以下。
MD計算をハンズオンで学習するための教育や、資金の少ない研究室でもマイクロ秒スケールのシミュレーションができるようにするため開発されたそうです。ありがたいですね!
1-2. 論文の紹介(Zenodo公開版)
査読済みの論文はオープンアクセスではありませんが、プレプリントをChemRxivやZenodoで公開してくださっています*3。計算の中身やコードの詳細というよりも、プロジェクトのモチベーションや大枠の構成の説明と、実行例の紹介といった内容になっています。
Making it rainの基本的構成は、① 計算パワーの必要なMD計算をGoogle ColabのGPUで実行し、② 入手力ファイルや設定ファイル、結果といったデータ保存・管理をGoogle Driveで行うものとなっています。
Google Colab、Google DriveともにGoogleアカウントがあれば無料で利用できますが、有償版ではより長時間・高容量の利用ができます。*4

計算時間の目安として、リゾチーム(lysozyme)を題材に1μsのシミュレーションを行った例が論文に書かれています。より高性能のGPUが使える有償版(Colab Pro)では、平均して約230ns/dayのシミュレーションが可能で、1マイクロ秒には6日あればOKだったそうです。
Colabには時間制限(有償版でも1日)があるため、Making it rainは長時間の計算を分割して実行できるように作られています。論文の例では、1マイクロ秒全体のトラジェクトリを20n秒ごと、50個のストライドに分割しています。各ストライドの終わりに、その時点での系の状態を保存したファイル(.xml)をOpenMMが出力してくれるので、出力ファイルを元に再度計算を実行することで連続したシミュレーションができる、とのことです。例では5回実行することで計1μsの結果を得ています。
1-3. ノートブックの種類
Making it rainではGoogle Colabで実行可能なノートブックが複数用意されており、それぞれ計算内容や計算方法が異なっているようです。論文では3つですが、現在はさらに3つアップデートされ、計6つのノートブックが利用可能です。
いずれもGitHubレポジトリのトップ(README)から利用可能で、こんな感じ。
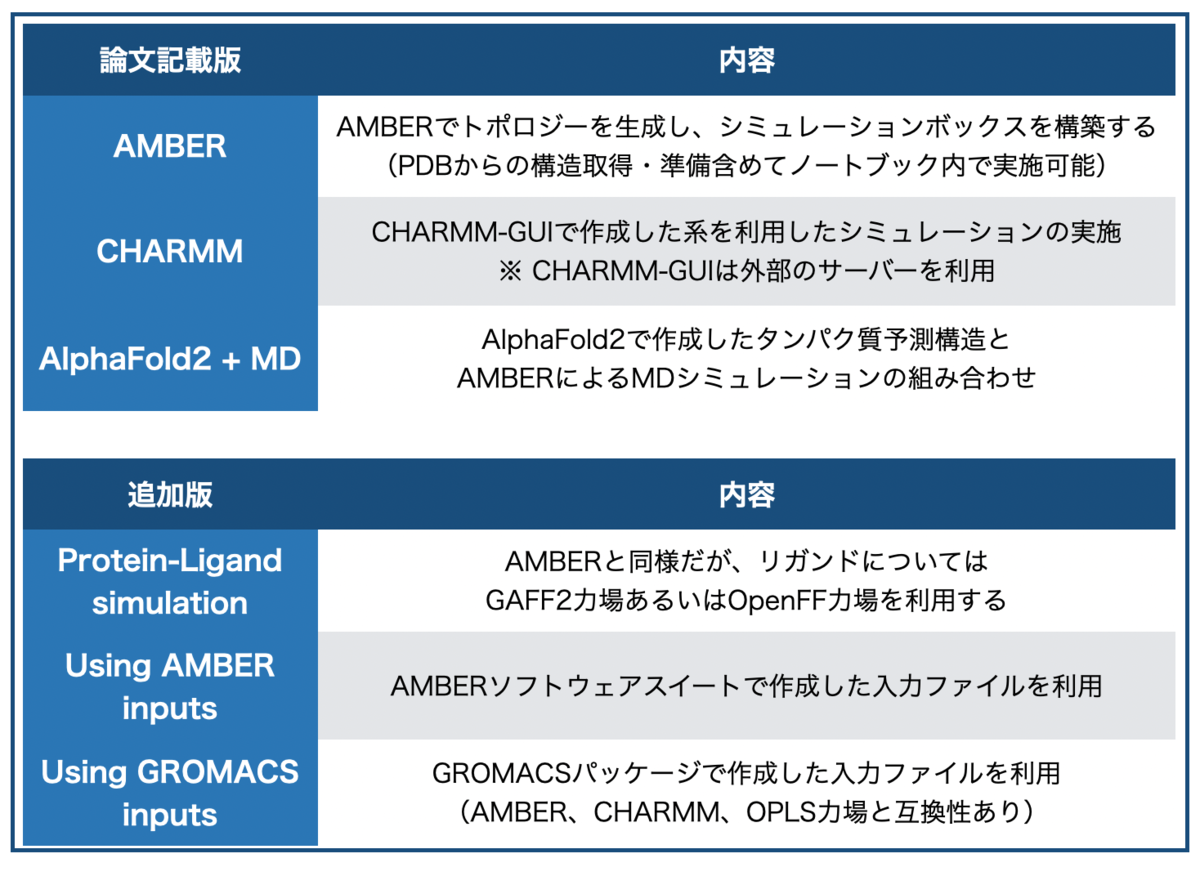
入力ファイルを作成するソフトウェアや利用する力場によってノートブックが分かれているようですが、MD計算を勉強したことがないので全く違いがわかりません。すみません。
よくわからないけどタンパク質モデリング(AlphaFold2+MD)やリガンドとの相互作用(Protein-ligand simulation)といったトピックもおさえてあるのが魅力的!!
以上、Making it rainの概要でした。
2. 使ってみた
2-1. AMBERノートブックの流れ
ざっとMaking it rainの目的が把握できたので早速遊んでみましょう!今回はノートブックのうち「AMBER」をそのままデフォルトで実行してみました。*5
AMBERノートブックの流れは以下のような感じでした。

「ノートブックを順番に操作するとどうなるか?」ビデオ形式のデモまで著者のPablo Arantesさんが用意してくださっています。ノートブックを開くとすぐに見られる(2分ちょっと)ので、流れを把握するのに便利です。親切!!*6
2-2. やってみた 〜MD計算の実行〜
「AMBERノートブック」のデフォルトの題材は「ニワトリ卵白由来のリゾチームの水中における挙動」のシミュレーションでした。MD計算実行部分がどんな感じだったかご紹介します。
まず、初期構造の準備です。PDBからPDB ID: 1AKIを取得しています。手持ちの構造が使いたければGoogle Driveに入れておけば参照できます。
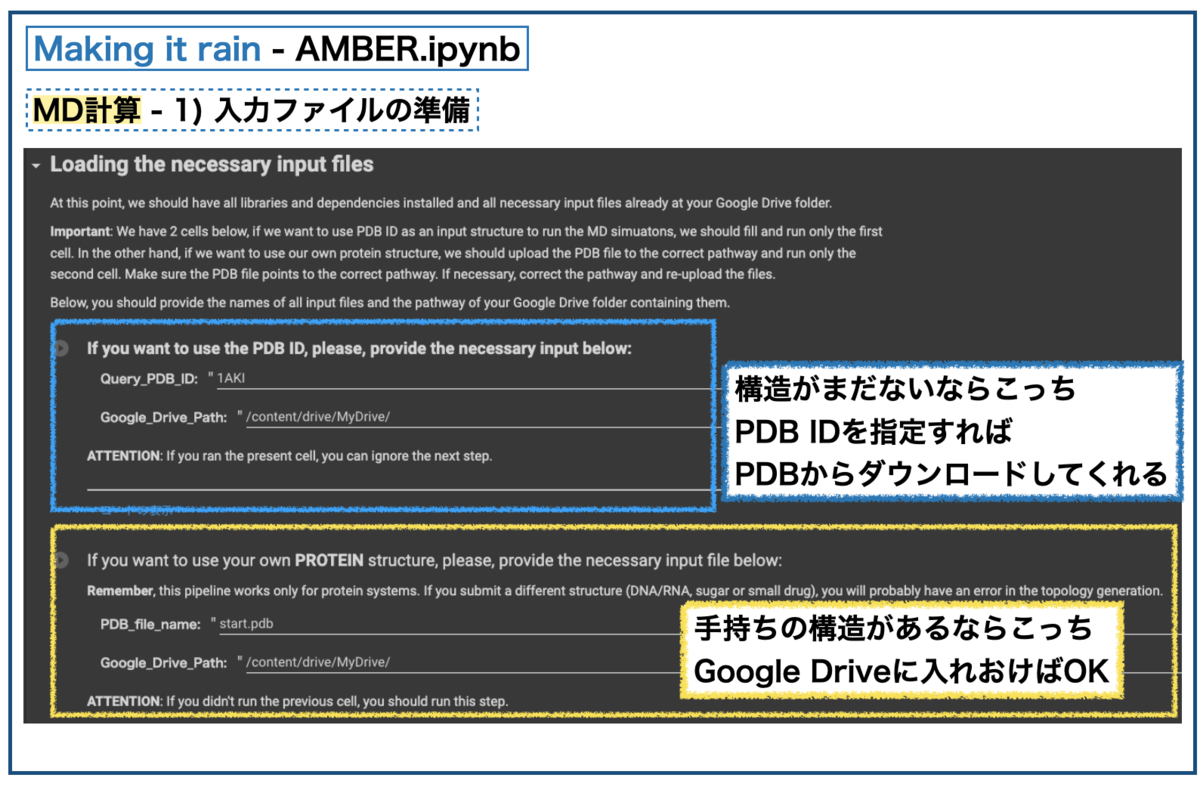
次にシミュレーションの系の設定です。水で満たされたボックスにタンパク質が一つ配置されます。さらにイオン(NaCl)を加えて系を中和しておきます。
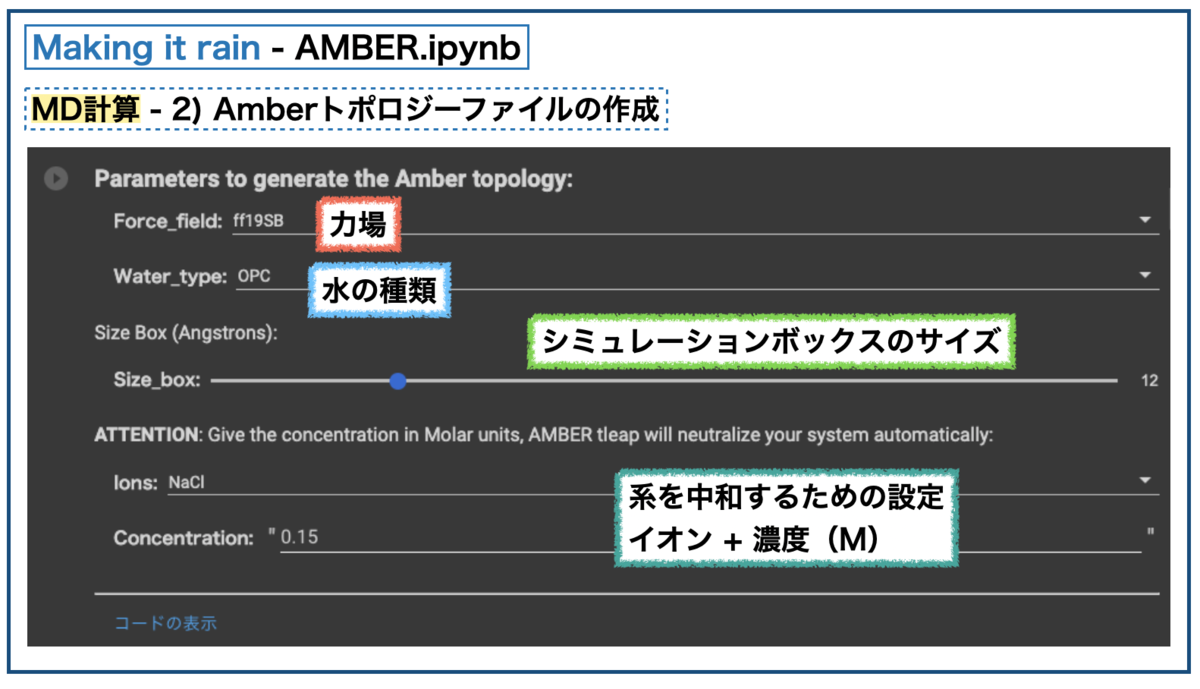
上がトポロジーファイルのパラメータ設定です。力場(force field。上ではff19SB)や水の種類、ボックスサイズ、イオンといった項目があります。これを元にMDシミュレーションのための系のパラメータがつくられます。
@Ag_smith先生の以下の記事に詳しいですが、AmberではtleapというモジュールでPDBファイルからトポロジーファイルを作成するそうです。実際にMaking it rainのコードを表示するとtleapを呼んでいます。
トポロジーファイル作成後、「シミュレーションの系がどのようにできたか?」、ノートブック上で3Dで可視化して確認できます。MD計算は時間が長いのできちんと準備できているか心配になりそうですが、ビジュアルで確認できると安心できていいですね!

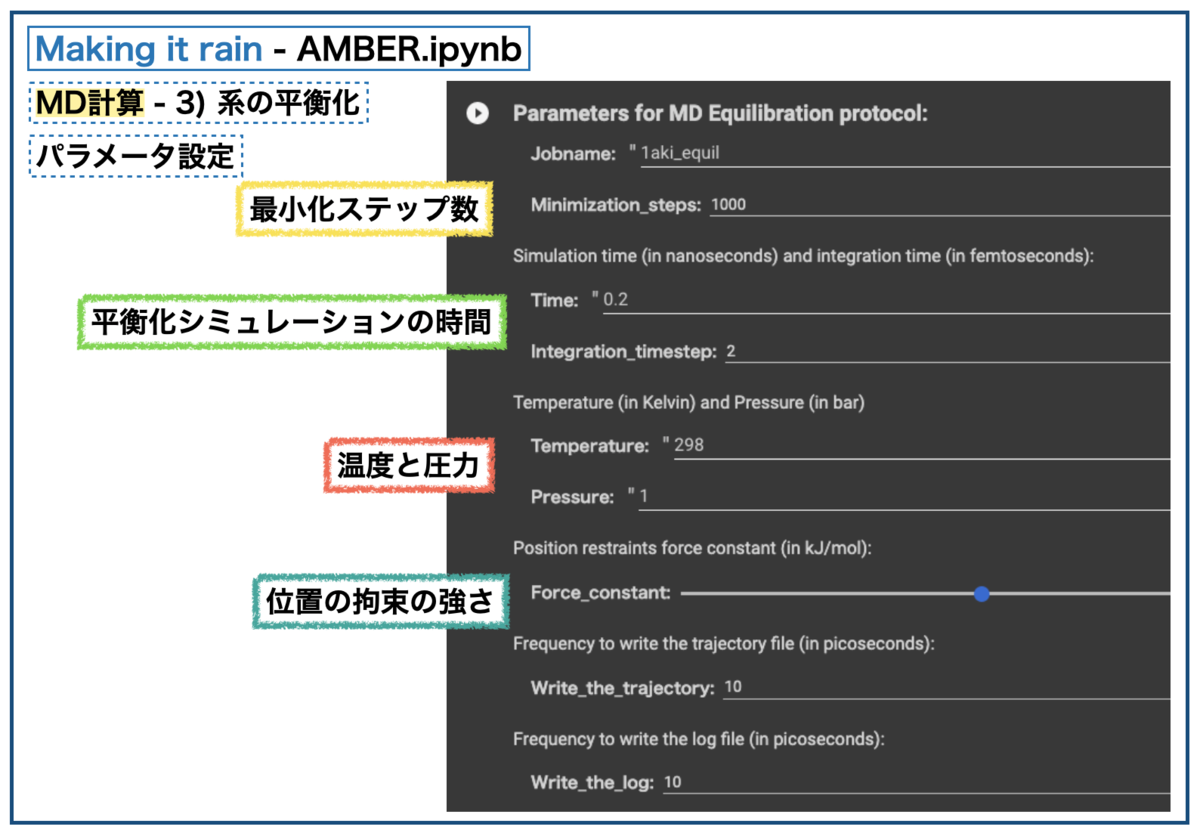
系が準備できたら平衡化(equilibration)を行います。MD計算ではシミュレーションの間、系の温度や圧力が一定の値を保つように計算を行うそうです。そのためにシミュレーションの本番前に短い計算で系をならしておくようです。*7
設定はこんな感じ。
先の@Ag_smith先生の記事から関連する解説を引用させていただきます。*8
ここからは水溶液中に存在するタンパク質を物理演算で動かしていくことになります。しかし、これまでの操作で作り出した初期座標では水溶液の様子を再現しているとは言えません。・・・(中略)・・・そこで、まずは系のエネルギーをある程度小さくしてから(=エネルギー最小化)、各原子に(温度に対するBoltzmann-distributionに従う範囲で)ランダムに速度を与え、徐々に系全体を設定温度まで引き上げていく(=平衡化)という予備動作を行うことで水溶液の状態に近づけていくという操作を行います。
①エネルギー最小化、②平衡化の順に実施するそうです。確かに、設定の最初にminimizationの文字が見えますね。
設定が終わったら平衡化シミュレーションの計算を実行します。NPTアンサンブルは物質の量(N)、圧力(P)、温度(T)が一定になるようにシミュレーションを行うそうです。「気温と大気圧に開放されているフラスコを用いた実験室条件に最も密接に対応している」らしい。。。(Wikipedia - 分子動力学法)

平衡化の長さは0.2nsとしていて、平均して約45ns/day(〜 0.03 ns/min)のスピードでシミュレーションできたようなので、計算時間は6分程度でした。"Time Remaining" で残り時間の目安がわかるのが親切ですね。
最後にシミュレーション本番(Production)を実行します。シミュレーションの長さはNumber_of_stridesとStride_Timeで指定し、2つの掛け算が得られる長さとなります。
例)100n秒シミュレーション = (ストライド数 10) x(各ストライドの長さ 10n秒)
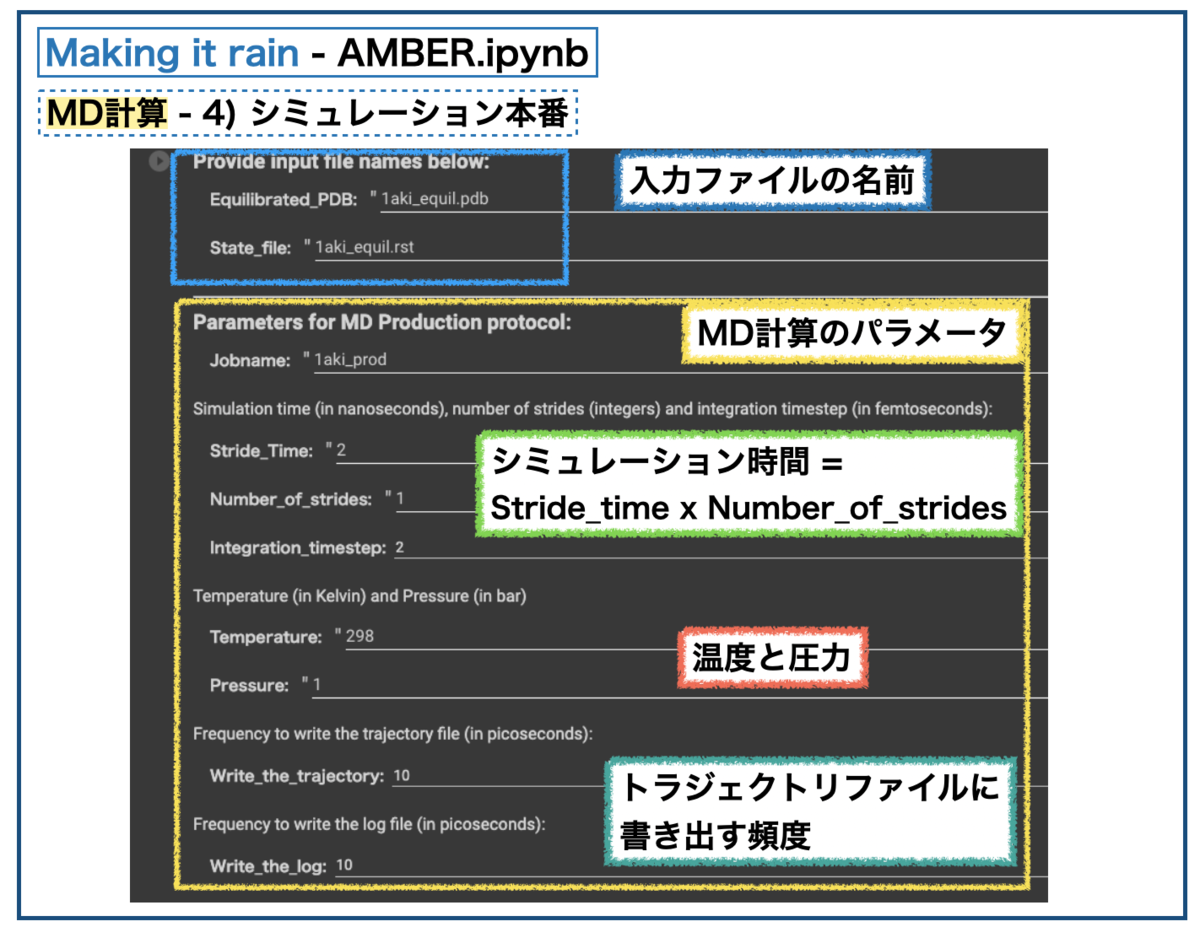
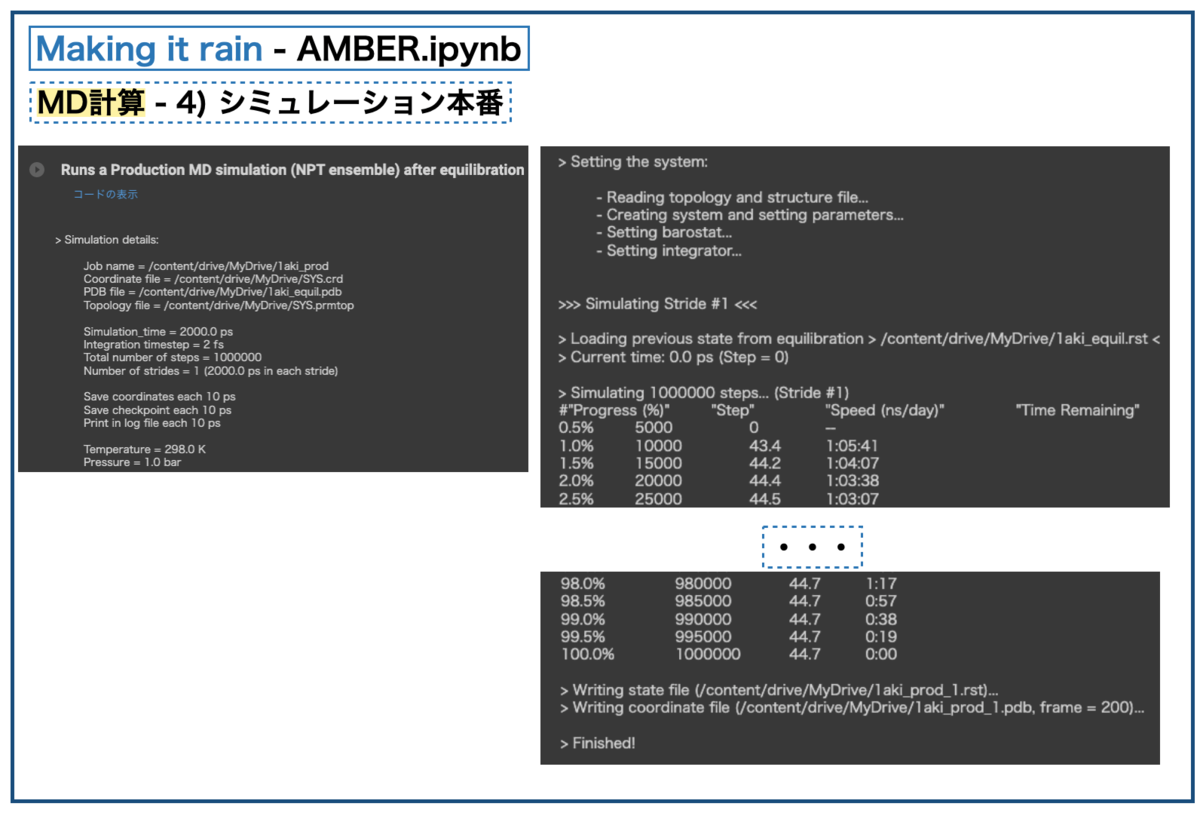
今回は全てデフォルトのまま2nsのシミュレーションを行いました。以下を実行し、計算時間は1時間ちょっとという感じでした。平衡化の10倍の長さなので、だいたい同じ感じのスピードで計算できた感じです。
MD計算が終わったら、シミュレーションの最後にトラジェクトリ(trajectory)ファイルを一つにまとめる操作を行います。
系に含まれる各原子の3次元座標の時間変化のことをトラジェクトリー(trajectory、軌跡)と呼ぶそうです*9。パラメータ設定で「トラジェクトリファイルに書き出す頻度」を10psとしたので、2nsのシミュレーションでは200個のファイルができることになります。これを一つのファイルにまとめます。
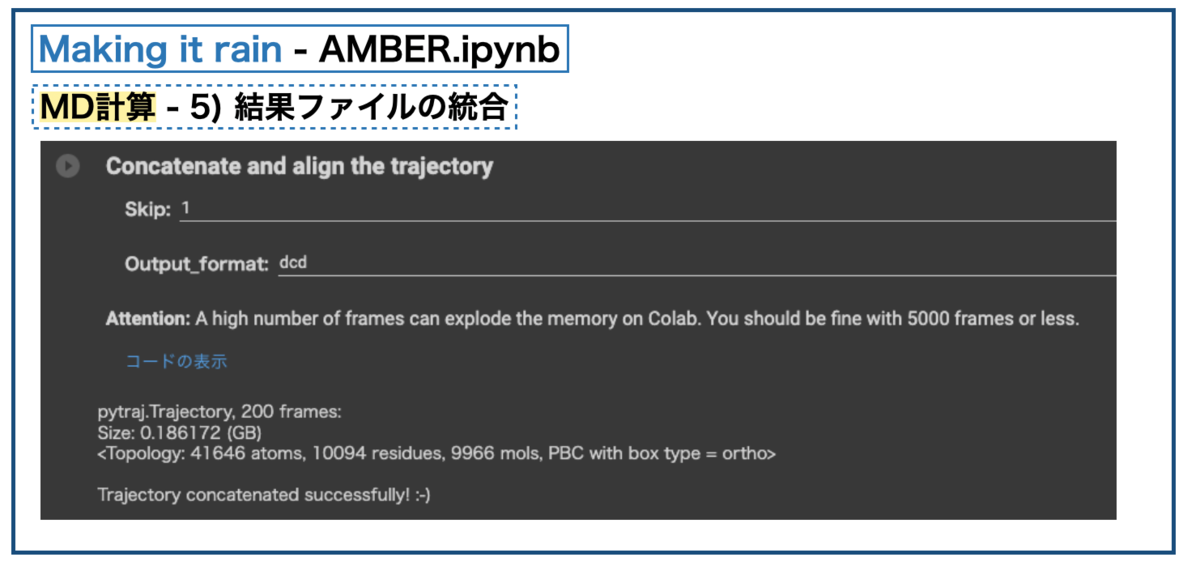
まとめると0.2GB弱でした。2nsで0.2GBなので1μsだと100GBくらいになりそうです。・・・でっかい。
2-3. やってみた 〜結果の解析〜
計算の実行とファイルの統合が終わったので、結果の解析部分を見てみましょう!
Making it rainのノートブックに用意されているので、引き続き実行結果をみていきます。
まずはトラジェクトリファイルを動画で見ます。「Load, view and check the trajectory」というセルを実行するだけ!
こんな感じ(以下はキャプチャのGIFです)

ピクピクしてる!!かわいい!
「動画もいいけど、もう少し定量的に結果を見たいよね!」というわけで、以下のような解析が用意されています。それぞれセルを実行するだけなので結果のプロットを貼っていきます。
まずは各アミノ酸残基のCα原子(中心炭素原子)についてこんな解析ができます。

①RMSD(根平均二乗変位, Root Mean Square Deviation)、②radius of gyration(慣性半径)、③RMSF(根平均二乗揺らぎ, Root Mean Square Fluctuation)です。①と②は時間に依存した変化と分布、③は残基ごとの値についてそれぞれプロットされています。
まったくどう解釈していいかわからない!!
①RMSDが0.5nsくらいまで増加傾向にあるのをみると、production前の平衡化が足りていなかったのでしょうか?
③RMSFでは末端以外にも残基40~50や70前後で少し大きくなっていますね。動画(GIF)で左側青色のループ部分に相当しそうなので、動きが大きそうな部位を数値化できてる感じでしょうか??。
残りの解析結果も貼っておきます。こんな感じ。

2D RMSD、PCA、Pearson's Cross Correlation (CC, ピアソン相関)。。。よくわかりませんでした。
結果の解析部分は以上です。
3. おわりに
以上、今回はGoogle Colab上でMD計算を実行できるMaking it rainと、実際に実行してみた結果のご紹介でした。
Making it rainではオープンソースのMD計算ソフトopenMMを利用しています。こちらはTeachOpenCADDでも使われているそうです。@iwatobipen先生が記事「Make mdtools for openmm」で紹介してくださっていました。*10
専門外の分野でも、オープンソースのツールがあったり使い方の解説があると「ちょっと遊んでみようかなー」となるので嬉しいですね!Making it rainはさらに低コストで高コストなMD計算を実施するフレームワークを提供してくれていて、とてもありがたいです。
残念ながら専門外すぎて、どういう設定で計算して、結果をどう評価していいのか全くわかりませんでした。不勉強ですみません。。。
ところで「Make it rain」という単語、直訳すると「雨を降らせる」ですが、Googleで検索すると強面のお兄さんたちと紙幣が舞う画像がでてきます。スラングで「大金をばら撒く、札束をまき散らす」といった意味があるとか。。。
クラウド(雲)コンピューティングから高価なデータの雨を降らせよう(たくさんお金がないとできないような計算をいっぱいやろう)なんて、意味が込められてたら面白いですね。
今回も色々と間違いが多そうです。ご指摘いただけると嬉しいです。
ではでは
追記:日本語化ノートブック
Making it rainのノートブックを日本語化したものをGitHubに入れておきました。ほとんどDeepL翻訳そのままで、動作確認もまだできていませんがご参考までにどうぞ。一応Colabでも動かせるはずです。
*1:引用したツイートと関連するMD計算の結果はDESRESのHPで公開されています(CC-4.0)。Molecular Dynamics Simulations Related to SARS-CoV-2。
*2:下の図はGitHubのFigureといらすとやさんのイラストを利用させていただいています。
*3:ChemRxiv上のライセンスはCC BY NC ND 4.0です。ZenodoはCC BY 4.0になっていますがサイトの設定か論文本文かどちらかわからなかったです。この記事ではZenodo公開版を参照させていただいています
*4:無償版/有償版の費用や性能は変わる可能性があるので適宜検索してください。Google Driveの有償版としてはGoogle Oneの情報を記載しました。
*5:何もわからないので一番上にあるやつをクリックしただけ。。。
*6:デモ動画はYouTubeにありますが「限定公開」設定なのでここにはリンクを貼らないでおきます。ノートブックをひらけばすぐわかります。
*7:全然違う話題ですけど、平衡化を最初に行うのベイズ推定のマルコフ連鎖モンテカルロ法(MCMC)使ったパラメータ推定の計算みたいですね。
*8:ただし引用記事はこの部分でAmberではなくGROMACSというソフトに切り替わっています
*9:参考:古明地 勇人, 上林 正巳, 長嶋 雲兵 生体分子の分子動力学シミュレーション(1)方法 J. Chem. Software, 2000(6)1–36
*10:TeachOpenCADD、インストールで力尽きて新しいトークトリアルの中身をまだ見られていないです。すみません。