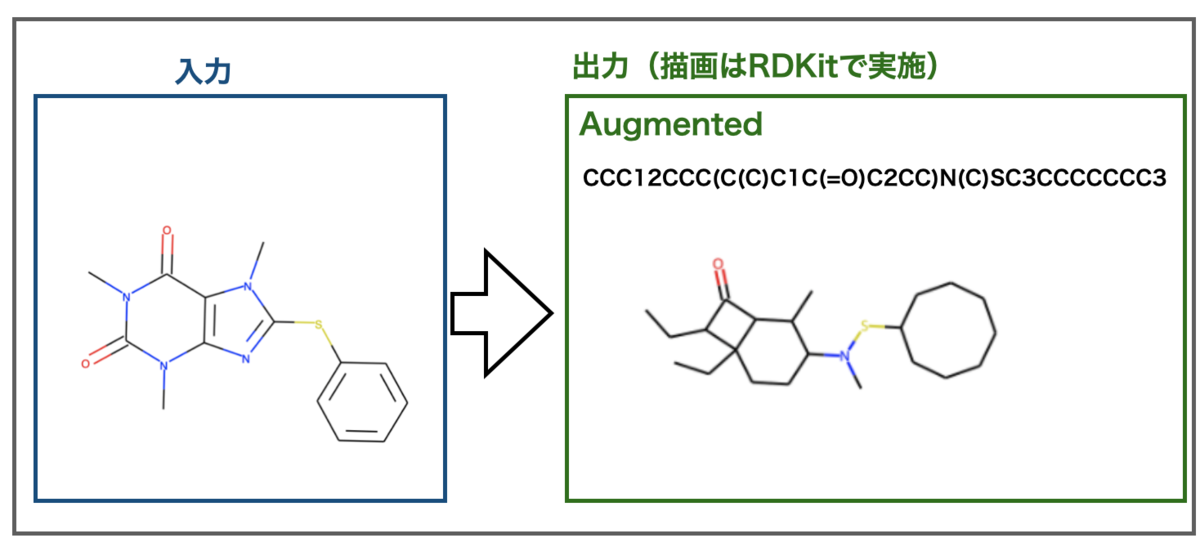
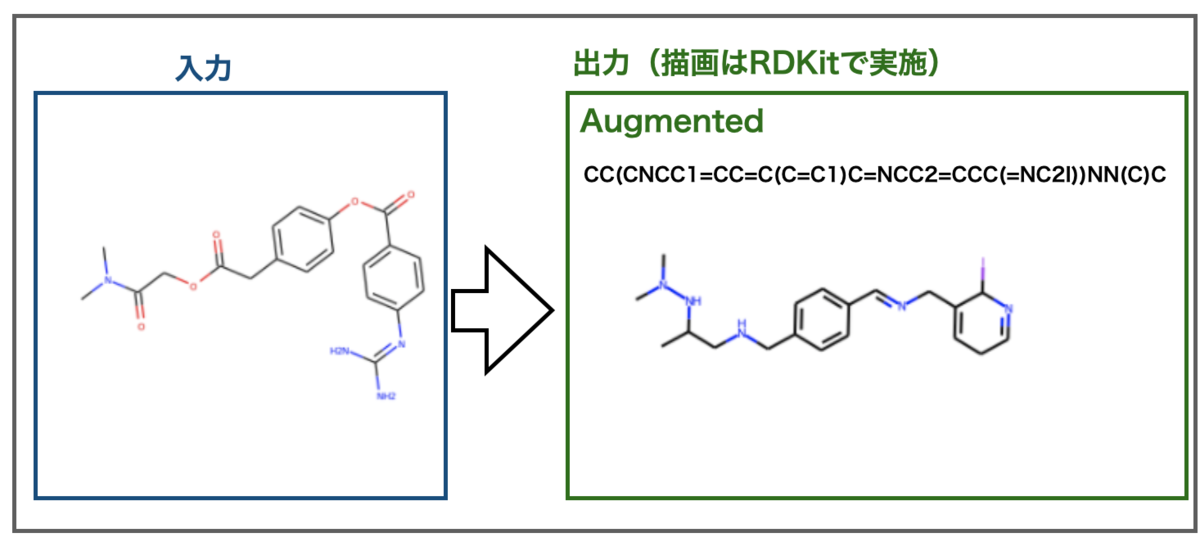
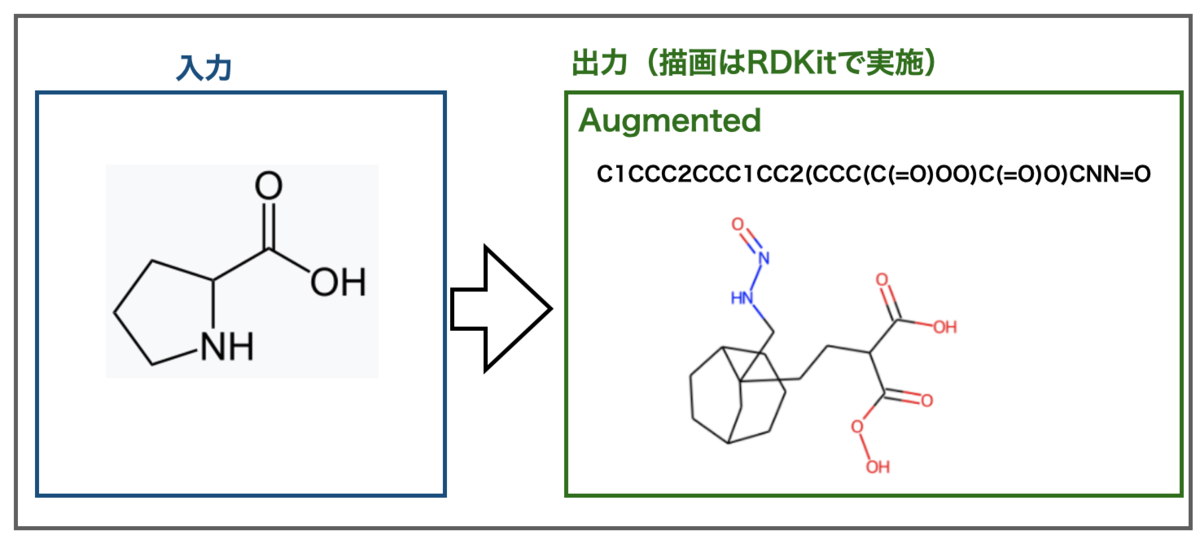
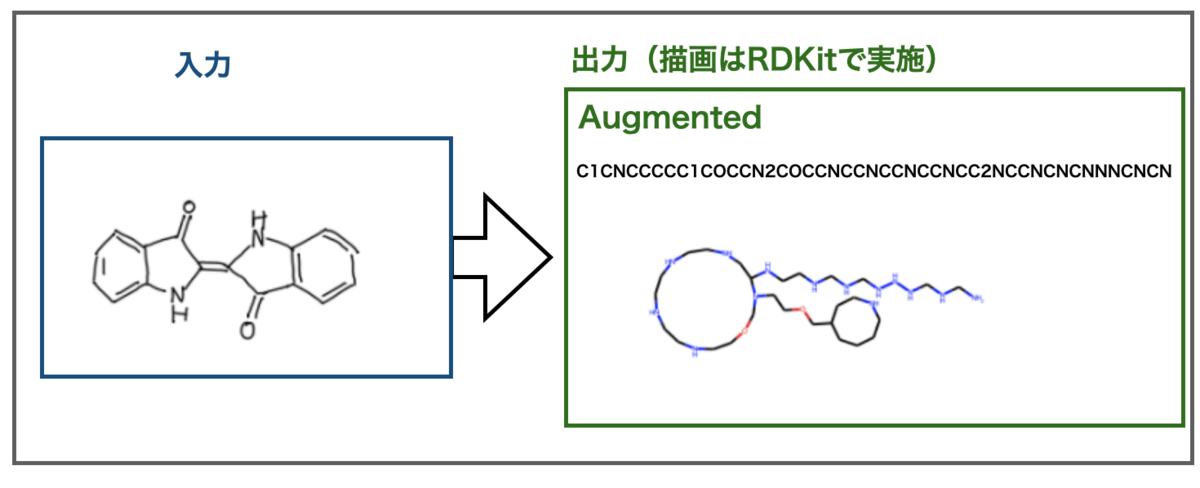
TeachOpenCADD 2021をインストールした話
大幅にアップデートされたTeachOpenCADD 2021がリリースされました!
とりあえずインストールした!!
ちょっとつまずいた!!
のでインストール方法のメモです。
- 1. TeachOpenCADDって?
- 2. TeachOpenCADD 2021
- 3. アップデートはコンテンツだけじゃない!Webサイトも充実!
- 4. まだまだ増えてるコンテンツ
- 5. オンライン上で遊ぼう!(インストール不要)
- 6. ローカル環境で動かしたい!〜インストール①〜
- 7. もっとローカル環境で動かしたい! 〜インストール②〜
- 8. おしまい
1. TeachOpenCADDって?
TeachOpenCADDは「計算機支援ドラッグデザイン(CADD, computer-aided drug design)の基本的なタスクをどのように実行すれば良いか?」学習・教育するためのプラットフォームで、ケモインフォマティクスや構造バイオインフォマティクスについて学ぶ事ができる教材です。
Volkamer研究室が主体となって開発されており、オープンソース(CC BY 4.0)で公開されています。
2019年に公開・論文発表されており、以前こちらのブログでも取り上げさせていただきました。
今回、大幅にアップデートされたTeachOpenCADD 2021が公開されましたので改めてご紹介します。
2. TeachOpenCADD 2021
TeachOpenCADDはCADDのトピックをそれぞれ取り上げたトークトリアル(talk + tutorial = talktorial)と呼ばれる教材で構成されています。
2019年版ではT1 ~ T10の10回のトークトリアルでしたが、2021年版はT22までと大幅に内容がアップデートされています。
ChemRxivに論文(preprint)が公開されています。
- Sydow D, Rodríguez-Guerra J, Kimber TB, Schaller D, Taylor CJ, Chen Y, et al. TeachOpenCADD 2021: Open Source and FAIR Python Pipelines to Assist in Structural Bioinformatics and Cheminformatics Research. ChemRxiv
Figure 1. をご覧いただけば扱われている内容がつかめると思います。

すごい!!
もう少し概要を知りたい方はVolkamer Labのブログ(TeachOpenCADD 2021 release is out!)や、RDKit UGM2021でのご発表などもご参照いただくと良いかも。。。
- UGM2021のライトニングトーク(関連発表は9分50秒くらいからスタート(10分弱))
- プレゼンテーション資料へのリンク Dominique Sydow - TeachOpenCADD update
3. アップデートはコンテンツだけじゃない!Webサイトも充実!
盛りだくさんにコンテンツがアップデートされたTeachOpenCADDですが、2021のアップデートはさらに豪華です!
格好いいWebサイトまでできました!!

素敵だと思ったのは「Talktorials by collection」。関連するトークトリアルをグループ分けしたものが確認できます。

構造バイオインフォマティクスってインスタ映えしそうですね。
4. まだまだ増えてるコンテンツ
TeachOpenCADD 2021はトークトリアル T22までのアップデートと書きましたが、実はコンテンツは継続的に開発されていてWebサイト上では最新コンテンツも確認できます。
Volkamer Labのブログ(TeachOpenCADD Kinase edition is out!, 2021.12.14)によると、キナーゼに焦点を当てたスペシャルエディションができたそう。
Webサイトでチェックだ!

新しいトークトリアル(T23 ~ T28)がアップデート済みです!ますます充実していますね!
5. オンライン上で遊ぼう!(インストール不要)
TeachOpenCADDで遊ぶにはオンライン上で実行する方法と、ローカル環境にインストールする方法の2通りがあります。
オンラインはBinderで実行する形式で、セットアップ不要で手間要らずです。TeachOpenCADDのGitHub上にある「lauch / binder」ボタンをクリックするだけ!
BinderのURLは「 https://mybinder.org/v2/gh/volkamerlab/TeachOpenCADD/master 」なので、毎回GitHubに行くのが面倒な方はブックマークしておくと便利かもしれません。
こんな感じでBinderが起動して環境を準備してくれます。

準備にちょっと時間がかかります(5~10分くらい?)が、ワンクリックで使えるお手軽さは最高です。
準備できるとJupyter Labが立ち上がります。こんなの。
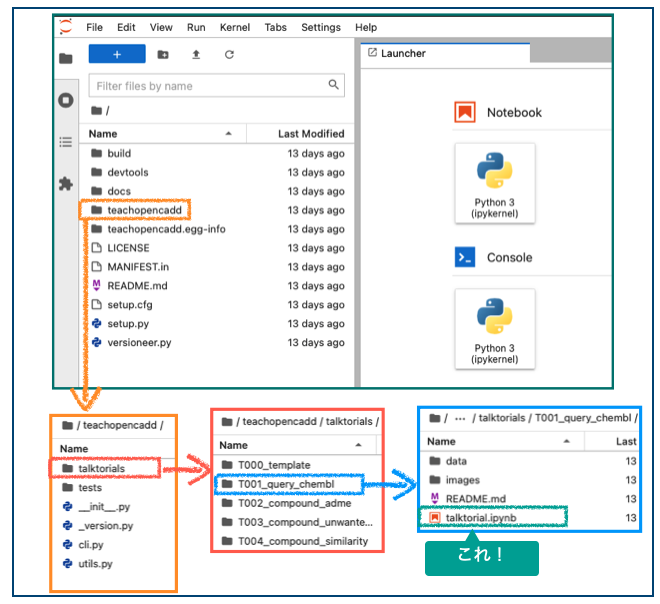
上図のように「teachopencaddディレクトリ → talktorialsディレクトリ → 見たいトークトリアルのディレクトリ(例:T001_query_chembl)」とたどって目的のノートブック(talktorkal.ipynb)を見つけます。
開くとWebサイト上で閲覧できるものと同じものが見られます。こんな感じ。
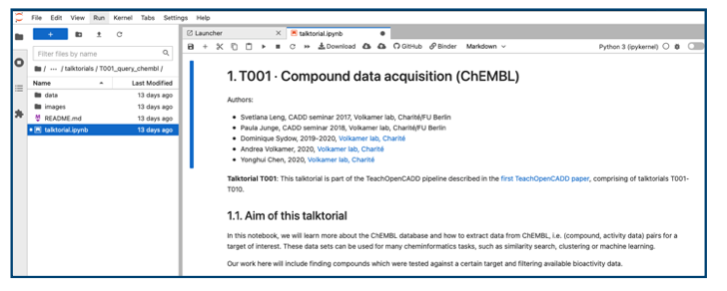
Webサイトとの違いは実際にノートブックのセルを実行できる事です。また編集できるので気になるコードを修正して結果の違いを見比べたり、試したい解析を追加したりと、いろんな遊び方ができそうです。
6. ローカル環境で動かしたい!〜インストール①〜
毎回Binderを待つのが嫌!オフラインでも試したい!という方はconda環境があればローカルにインストールすることもできます。
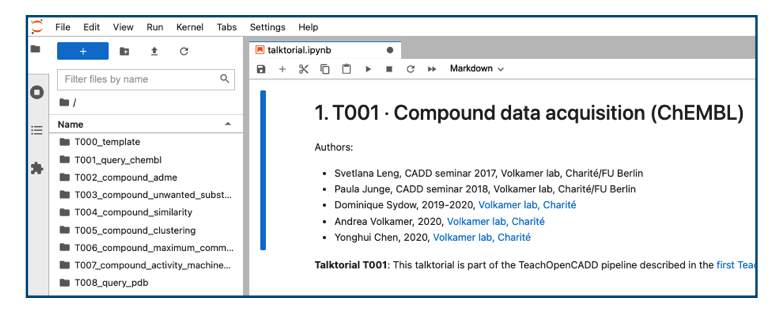
WebサイトのRun locallyページにインストール方法が書かれています。
Condaパッケージからのインストールは下記で、mambaを使った方法が書かれています。
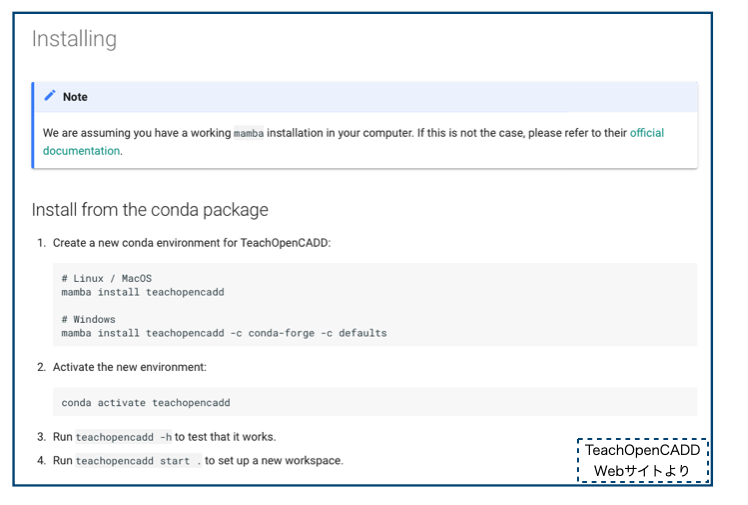
mambaは「C++で実装された高速なcondaパッケージマネージャー」だそうです*1。conda環境を使われているなら、mambaを追加インストールしておけば諸々のパッケージインストールが速くなるみたいです。(mambaオフィシャルドキュメント)
mambaは以下でインストールできます。私はMacなのでターミナルで以下を実行しました。
conda install mamba -c conda-forge
インストールできたら今まで「conda install ~~~」としてきたのを「mamba install ~~~」とするだけでOKです。
なお、TeachOpenCADDのインストール方法では新しい仮想環境をつくる工程が省略されているようです。通常通りconda create -n <仮想環境名>とすればOKですが、仮想環境の作成はmambaでもできるそうです。
やってみた!
# 仮想環境の作成 mamba create -n teachopencadd
できた!

上図の通りできた仮想環境(teachopencadd)のアクティベート、ディアクティベートの方法はcondaと変わらず同じコマンドです。
なお、mambaで作成した仮想環境もconda info -eで確認できる環境一覧に出てきますし、削除したくなったら以下でOKです。
conda remove -n teachopencadd --all
仮想環境ができたら、アクティベートしてTeachOpen CADDをインストールします。(Webサイトの順番だとできない気がするので逆にしてます)
# 仮想環境のアクティベート conda activate teachopencadd # TeachOpenCADDのインストール mamba install teachopencadd
インストールできたら以下のコマンドを実行しましょう!
teachopencadd start .
カレントディレクトリ(.)に新しいワークスペースが作成され、ターミナルにこんなのが表示されます。
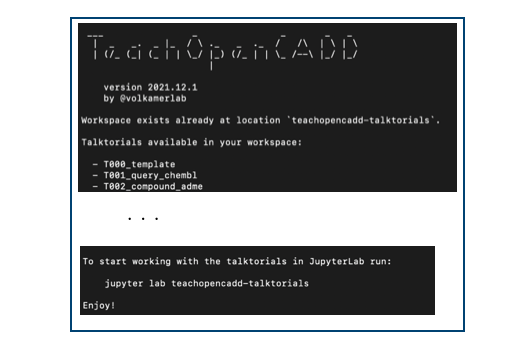
ロゴがかわいいですね。使えるトークトリアルの一覧とJupyterLabの起動コマンドが書かれています。
jupyter lab teachopencadd-talktorials
指示通り上記コマンドを実行すればBinderで確認したのと同じページがローカルで立ち上がります。
無事インストールできました!
なお、ワークスペースを作成する場所を指定したい時はteachopencadd start .コマンドのピリオドの部分にお好みのパスを入れてあげてください。
7. もっとローカル環境で動かしたい! 〜インストール②〜
「リリースを待ちきれない!」という方や、「自分で新しいトークトリアルを作るぜ!」という方は最新の開発版をインストールされるのも良いかもしれません。
一応ご紹介しておきます。
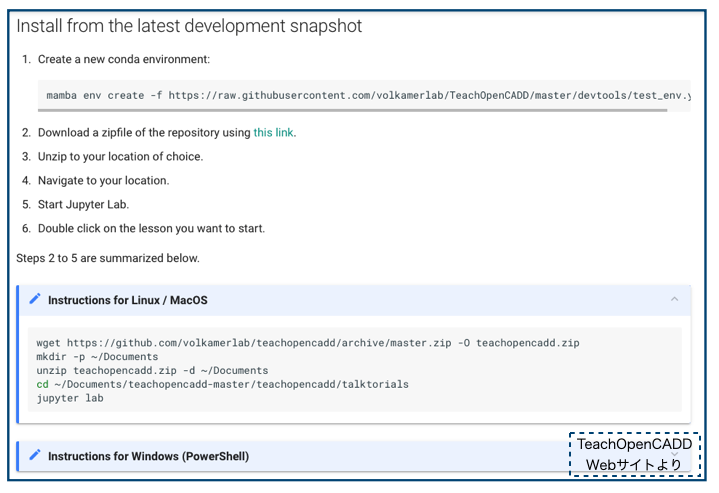
以下のコマンドでGitHubのTeachOpenCADDレポジトリに用意されているテスト環境用のtest_env.ymlを利用して仮想環境teachopencaddが作成されます。
mamba env create -f https://raw.githubusercontent.com/volkamerlab/TeachOpenCADD/master/devtools/test_env.yml
次に、以下でGitHubのレポジトリが作業ディレクトリ内にZIPファイルとしてダウンロードされます。
wget https://github.com/volkamerlab/teachopencadd/archive/master.zip -O teachopencadd.zip
unzipで解凍すればOKですが、以下では新しくDocumentsディレクトリを作ってその中に解凍しています。
# 作業ディレクトリ(test)の下にDocumentsディレクトリを作成 mkdir -p ./Documents # Documentsディレクトリ内にファイルを解凍 unzip teachopencadd.zip -d ./Documents
これでteachopencadd-masterディレクトリができました。この中のtalktorialsディレクトリに各トークトリアルのディレクトリが入っています。
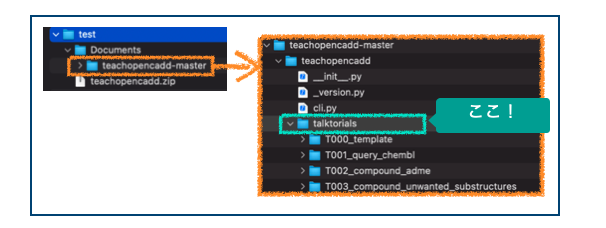
あとはtalktorialsディレクトリに移動してJupyterLabを立ち上げればおしまいです!仮想環境(teachopencadd)のアクティベートも忘れずに!
# talktorialsディレクトリに移動 cd ./Documents/teachopencadd-master/teachopencadd/talktorials # 仮想環境のアクティベート conda activate teachopencadd # JupyterLabの立ち上げ jupyter lab
mamba installしたときと同じJupyterLab画面が立ち上がりました!
先に見たインストール方法①ではワークスペース作成コマンドの実行があり、talktorialsフォルダができました。開発環境版を使う場合はワークスペース作成の代わりに、GitHubレポジトリを丸ごと持ってきてtalktorialsディレクトリをそのまま利用する、という違いがあるようです。
8. おしまい
以上、新しくパワーアップしたTeachOpenCADDについて簡単な紹介とインストール方法のご紹介でした。
コンテンツの大幅なアップデートだけでなく、Webサイトもとても見やすくなっていて素晴らしいですね!
Webサイトに書かれているインストール方法そのままだと、どうも私の環境(mac)ではうまくいかなかったので、ちょっとだけ手順をかえたものを記事にしてみました。色々と理解しないまま行っているので間違っているかもしれません。ご指摘いただけると嬉しいです。
なお、2021年版ではアップデートがなかったようですが、2019年版はKNIME版もありました。
- TeachOpenCADD-KNIME: A Teaching Platform for Computer-Aided Drug Design Using KNIME Workflows
J. Chem. Inf. Model. 2019, 59, 10, 4083–4086
こちらナイメストさんがnote上で使い方を解説してくださっています。
KNIMEの各ノードについて初歩から丁寧に説明してくださっていて、ほとんどKNIMEを使ったことのない私でも「ぽちぽちクリックしてたら動いたけど、背景ではこんなことが行われてるのねー」ととても勉強になるのでおすすめです。
正直、コードをみてもよくわからないのでKNIME版のアップデートが待ち遠しいです!!
なおなお、ついでのついでに皆様TalktorialsフォルダのトップがいずれもT000_templateとなっていることにお気づきになられましたか???
明日のTeachOpenCADDを作るのは君だ!!!
おしまい。
医薬品と回転異性体 〜実例とちょっと計算の検証〜
こちらは創薬アドベントカレンダーに間に合わなかった記事です。毎度、初歩の初歩の記事で恐縮ですが折角なのでここに供養します。
さて、クイズ!以下の4つの医薬品!構造も作用機序も全然違いますが、ある共通点があります!一体なんでしょうか??
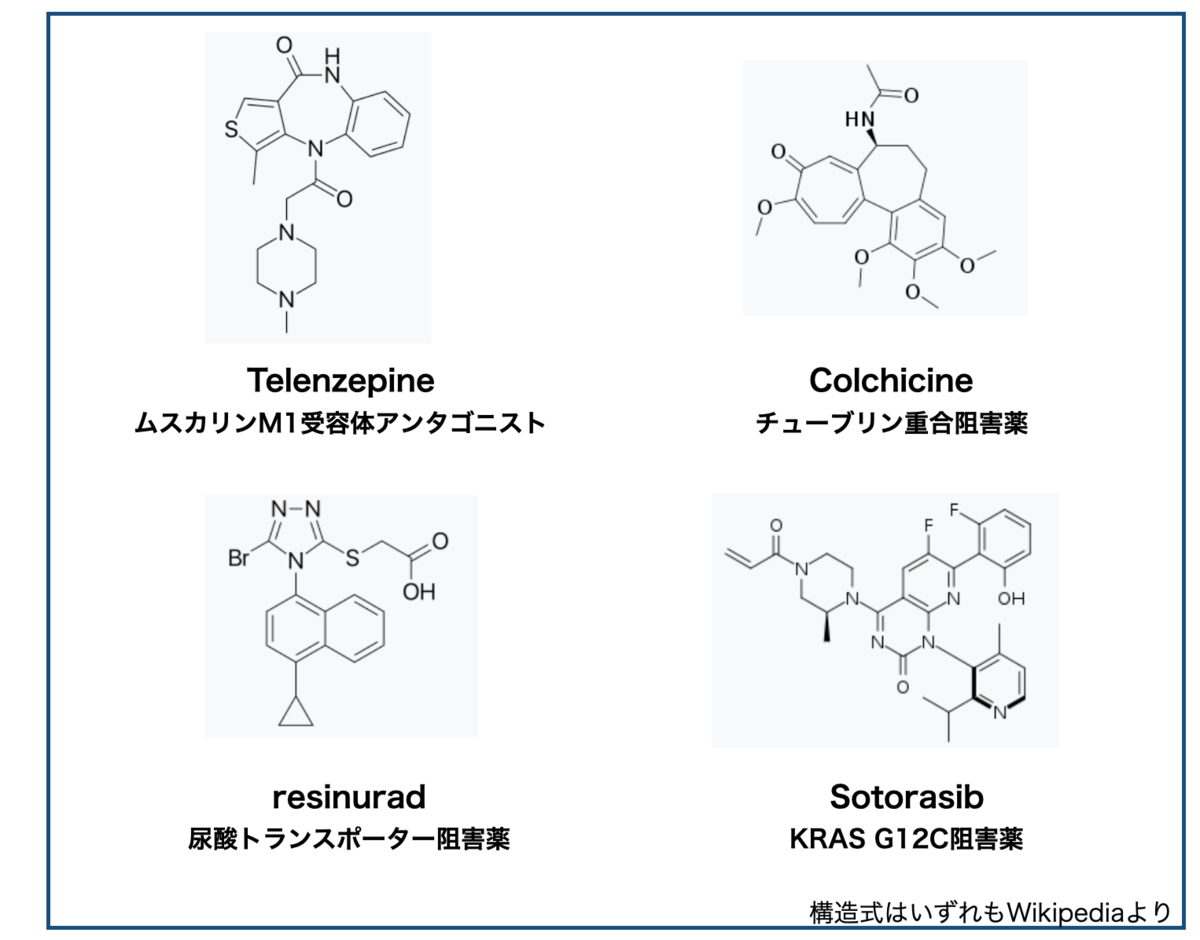
答え・・・「回転異性体(アトロプアイソマー)をもつ医薬品」。タイトルでネタバレしてましたけどね!
正直、私は構造式を見ただけではどこに回転異性が潜んでいるか分かりません。ということで、回転異性体判別力(?)をあげるべくエネルギー変化をシミュレーションして遊んでみたいと思います。
1. 回転異性と医薬品
まずは基本的な背景知識のおさらいです。
1-1. 回転異性の復習
低分子医薬品において不斉(キラリティー)は重要な要素です。2021年ノーベル化学賞「不斉有機触媒」の解説でも、不斉合成の重要性の例として医薬品が引き合いに出されていました。
この不斉の一つ、軸不斉は「分子が不斉中心を持たないが、キラリティー軸を有するキラリティーの特別な形式の一つ」です(wikipedia)。
軸不斉の例として回転異性(アトロプ異性)があり、回転異性を持つ化合物の例としては野依先生が合成方法を確立されたBINAPがあります*1。
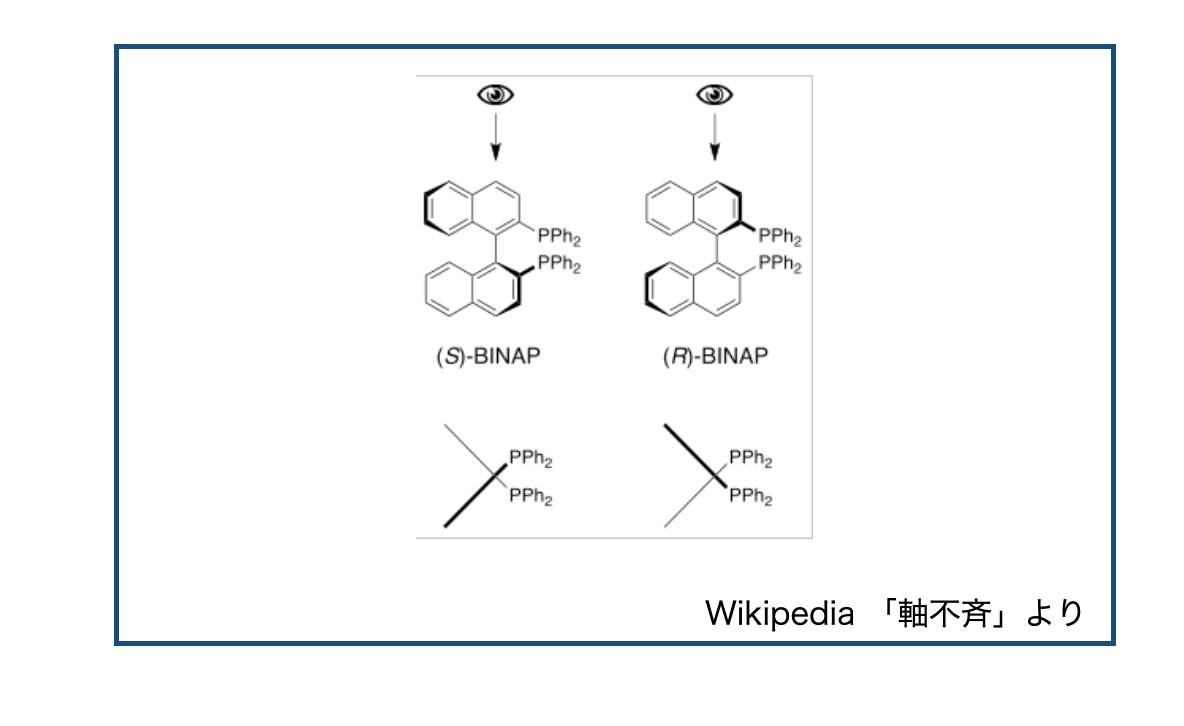
要するに、立体障害が大きくて自由回転できないから鏡像異性体ができちゃうぜってやつです。*2
1-2. 冒頭の例ではどこ?
「4つ異なる置換基がついた炭素」みたいな分かりやすい不斉中心がないので、回転異性体の有無はパッと見では分かりにくい気がします。
冒頭に挙げたそれぞれの化合物の不斉に関する軸はここです。
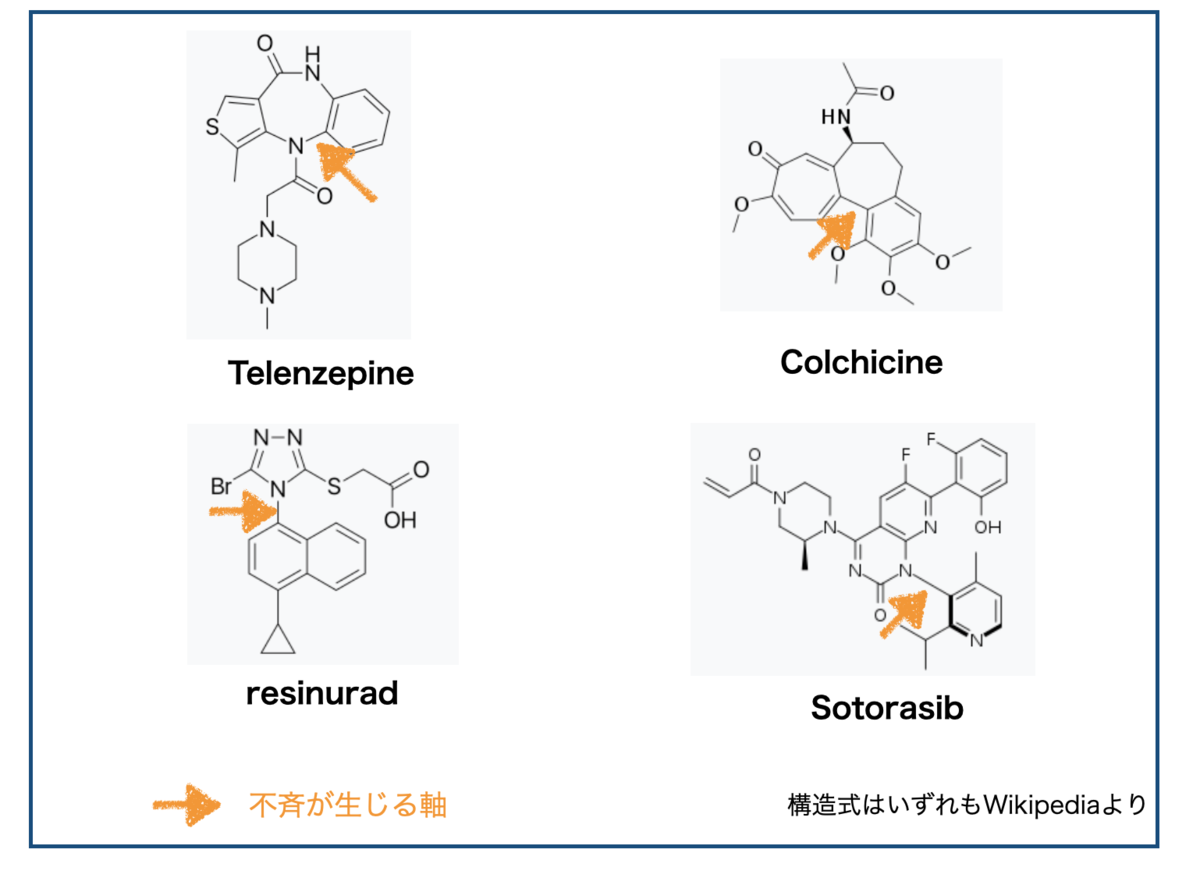
いずれも(ヘテロ)アリールに結合する箇所です。BINAPと見比べると雰囲気がわかってきました。
ちなみにWikipediaによるとTelenzepineは(+)-isomerと(-)-isomerでムスカリン受容体に対する活性に500倍以上差があるそうです(ラット大脳皮質)。鏡像異性体ってやっぱり生理活性に重要なんですね。
1-3. 回転異性体の安定性と分類
「回転異性体で活性差が大きいなら、良い方のだけ医薬品にすれば良いんじゃない?」となりますが、そんなに簡単ではないそうです。なぜなら、回転異性体の中には安定に分離できるものとできないものがあるからです。
回転異性体は軸周りの回転障壁の高さに由来します。障壁が十分に高ければ、異性体間での変換が十分に遅い(実質、変換しないとみなせる)ので、分離してそれぞれ別の化合物として扱う事ができます。一方で、障壁が中くらいであれば異性体を分離したところで、しばらくするとまた混ざってしまうので分離する意味がありません。
厳しい条件をクリアして医薬品として開発・臨床応用するためには、その化合物の均一性の保証も大事な要素です。生理活性も異なる2つの異性体が作るたびにバラバラに混ざってしまったら困ってしまいます。
以下の様に、回転障壁の高さに応じて回転異性体の安定性はざっと3つに分類できるそうです。(障壁のエネルギーは目安)*3*4
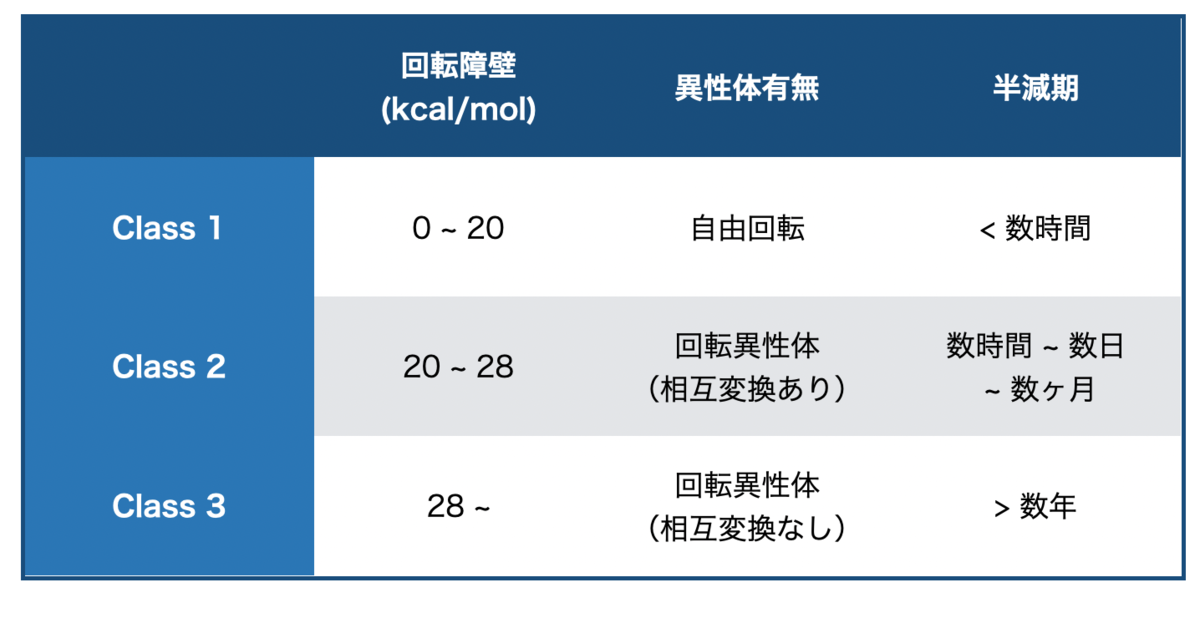
エネルギーの低いClass 1であれば、そもそも区別できるほど安定な回転異性体が生じないので一つの化合物として取りあつかえます。また、障壁が高いClass 3であれば年単位で安定に取り扱えます。面倒なのが中間のClass 2で、場合により、単体あるいは混合物として取り扱うようです。
1-4. 回転異性体への対処例
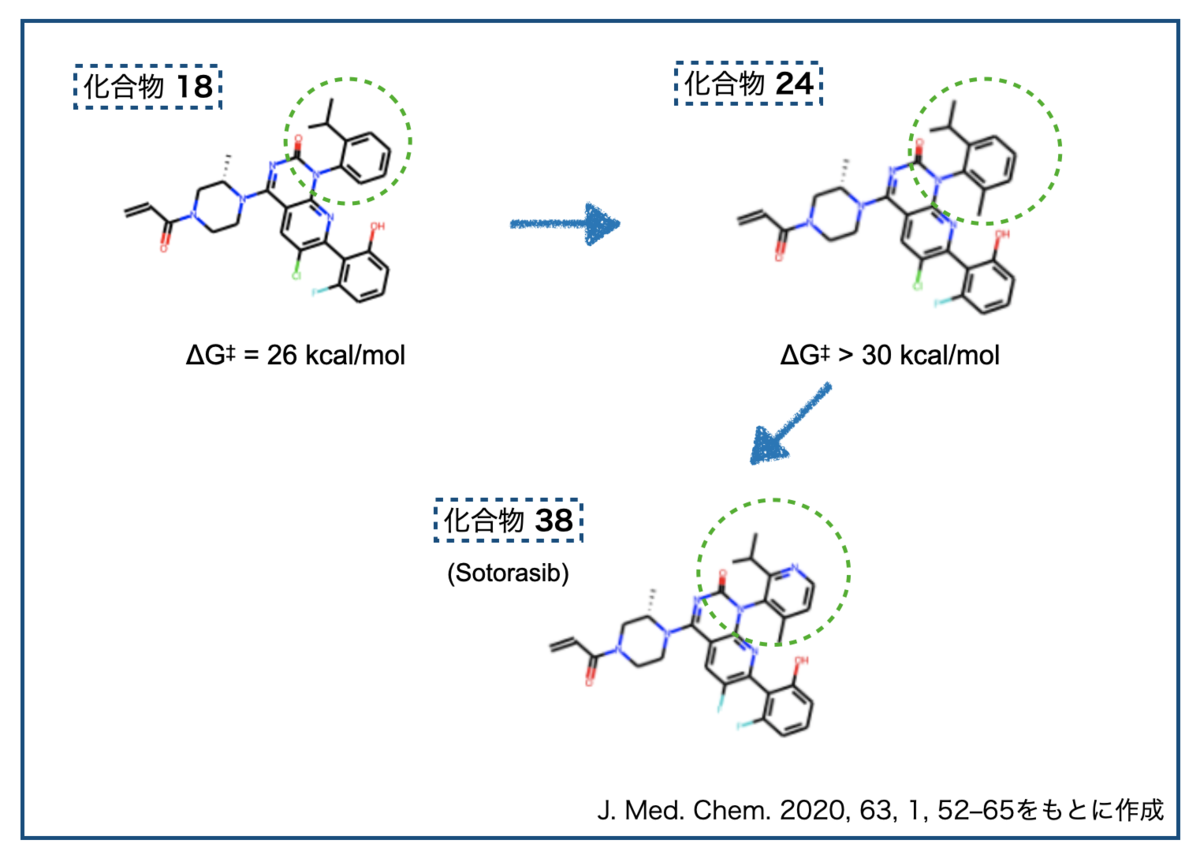
実際のところ、研究過程で回転異性体がでてきたらどうするのでしょうか?今年FDAに承認されたAmgen社のKras G12C阻害薬 Sotorasibの発見に至る経緯が面白いので参照しておきます。
- Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors
J. Med. Chem. 2020, 63, 1, 52–65
オープンで読めます!*5
研究の途中で見つかった化合物 18は有望な性質を持っていたものの、回転異性体がみつかりました。障壁は26 kcal/molで、25℃での半減期が8日。先の分類でいうとClass 2にあたりそうです。*6

回転異性体に対する戦略として著者らがあげているのは以下の3つです。
- ① もっと回りにくくして安定な異性体を分離できる様にする(Class 3にする)
- ② 逆に回りやすくして回転異性体が生じない様にする (Class 1にする)
- ③ 分子を対称形にして、回転しなくても鏡像異性体にならないようにする
検討の結果、戦略①がうまくいったそうです。軸不斉近傍にメチル基を導入することで回転障壁が30 kcal/molより大きくとなった化合物 24を見出しています。ここからさらに発展させて最終的にSotorasibを見出しています。
メチル基一つで回転異性という現象を制御してしまうの格好いいですね!承認された医薬品の構造式だけを見ても分かりませんが、そこに至る経緯を辿ると科学者の戦いが垣間見えて楽しいです!
2. 2面角とエネルギー変化
前置きが長くなりました。
構造式だけを眺めるだけではイマイチよくわからない回転異性体ですが、軸周りに回転させたエネルギー変化をたどれば感触がつかめるかもしれません。オープンソースのツールを使ってシミュレーションしてみましょう!
試す手法は以下の2つです。
- ① RDKitのコンフォマー生成による解析(Molecular Mechanics)
- ② xTBの2面角スキャンによる解析(Semiempirical Extended Tight-Binding)
以下の2つの記事で解説してくださっている内容を参考にさせていただいています。というよりほぼコピペです。ごめんなさい。
- 参考記事1:化学の新しいカタチ:RDKitを用いて制約付きで立体構造を生成する
- 参考記事2:Spinning coral : Torsion angle scan with rdkit & xtb
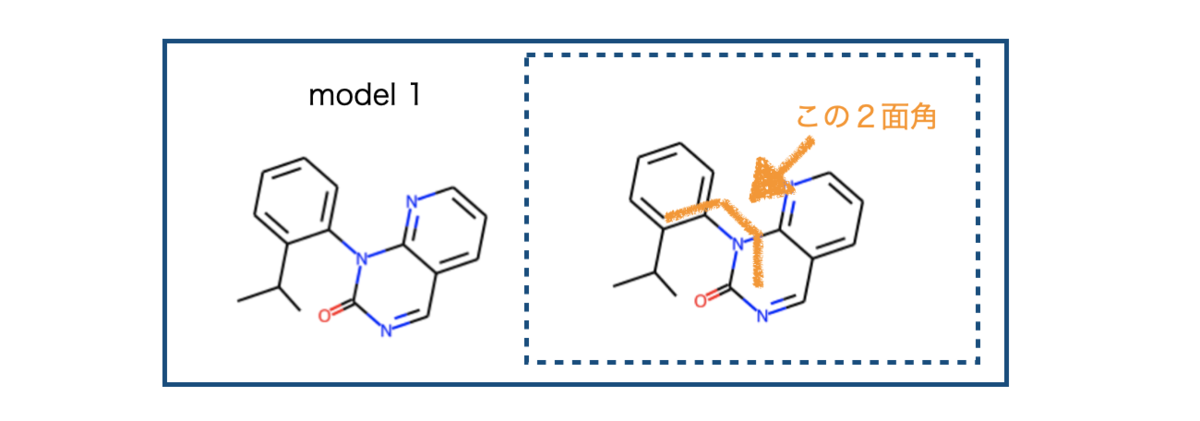
まず試すのは以下のモデル化合物です。先のJ .Med. Chem. の文献を参考にしています。
それでは2面角に依存してエネルギーがどのように変わるか? 眺めてみましょう!
2-1. モデル化合物 1の描画とatom indexの確認
まずはモデル化合物 1を描画して必要な情報を確認します。Molオブジェクトを作成し、考慮する二面角のatom indexをチェックします。
from rdkit import Chem from rdkit.Chem import AllChem, Draw # モデル構造作成 model_1 = Chem.MolFromSmiles("CC(C)c1ccccc1n3c(=O)ncc2cccnc23") # 水素原子付加 model_1H = AllChem.AddHs(model_1) Draw.MolToImage(model_1H)
RDKitのブログ記事より、drawMolDraw2DでaddAtomIndices=Trueとすることでatom indexを確認できます。*7
from rdkit.Chem.Draw import rdMolDraw2D from IPython.display import SVG d2d = rdMolDraw2D.MolDraw2DSVG(350,300) d2d.drawOptions().addAtomIndices=True d2d.DrawMolecule(model_1H) d2d.FinishDrawing() SVG(d2d.GetDrawingText())
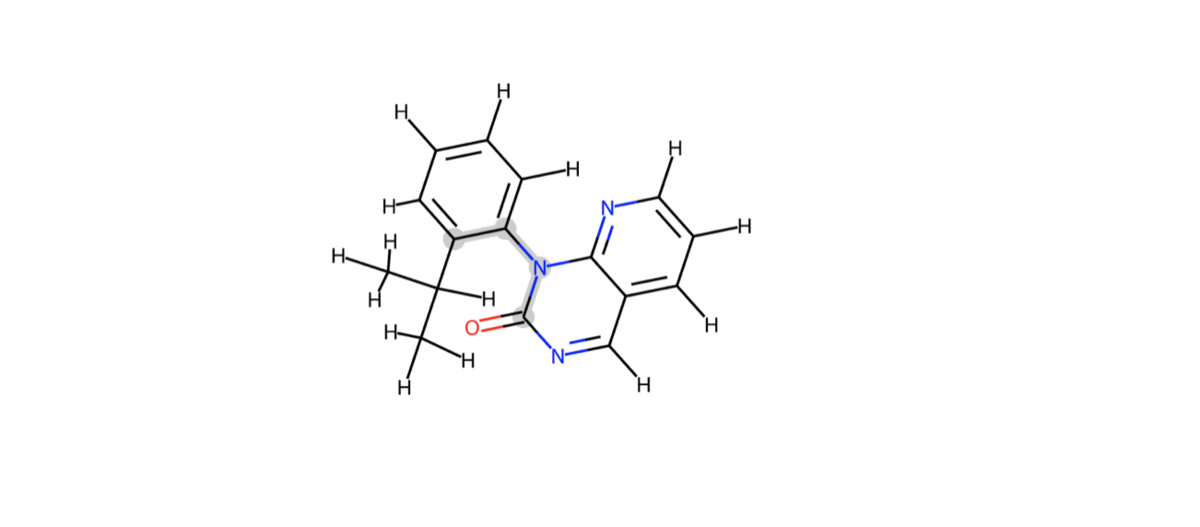
イソプロピル基の根元から軸周りのatom indexを今回の標的とします。小さくて見づらいですが3, 8, 9, 10が目的の2面角を定義する原子のatom indexです。
ハイライトして描画してみます(highlightAtoms)。
cp = Chem.Mol(model_1H) d2d = rdMolDraw2D.MolDraw2DSVG(350,300) d2d.drawOptions().setHighlightColour((0.8,0.8,0.8)) d2d.DrawMolecule(cp,highlightAtoms=[3, 8, 9, 10]) d2d.FinishDrawing() SVG(d2d.GetDrawingText())
結合(bond index)についても同じことはできます(addBondIndices=True)。見やすさのため水素原子を付加せずに確認します。
d2d = rdMolDraw2D.MolDraw2DSVG(350,300) d2d.drawOptions().addBondIndices=True d2d.DrawMolecule(model_1) d2d.FinishDrawing() SVG(d2d.GetDrawingText())
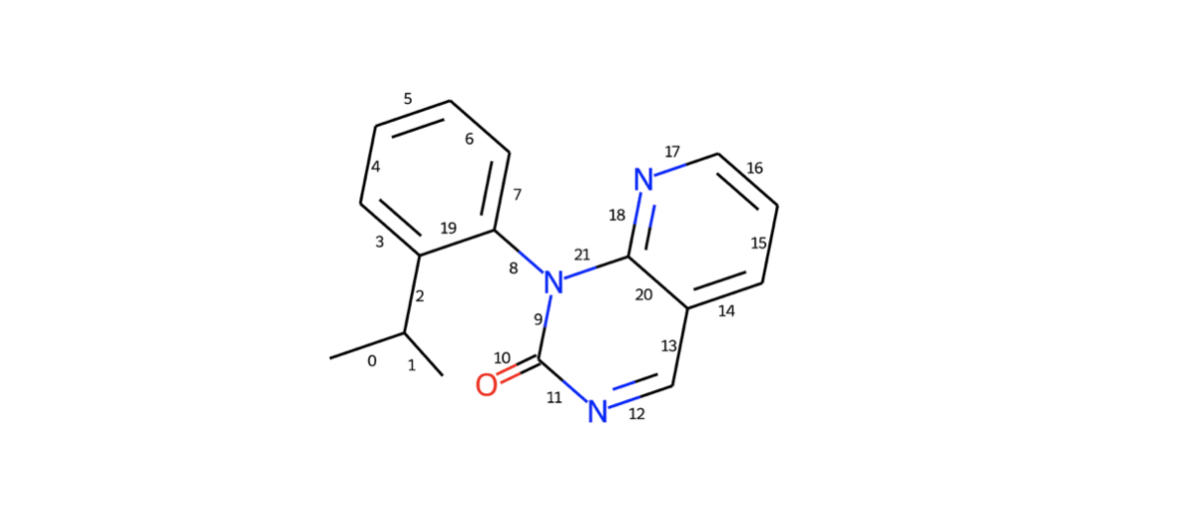
着目部位のbond indexは19, 8, 9でした。
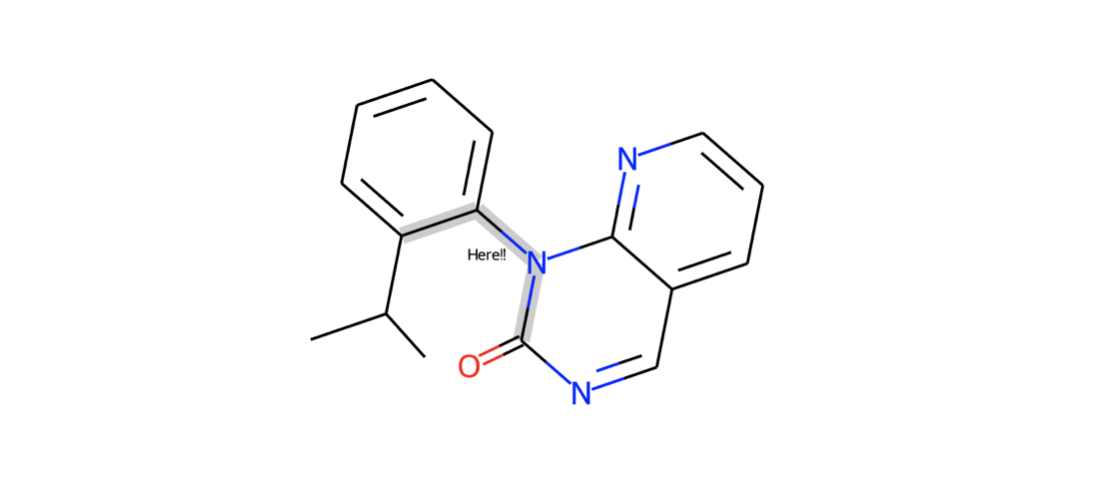
結合にコメントをつけて描画する事もできます(bondNote)。こんな感じ。
cp = Chem.Mol(model_1) cp.GetBondWithIdx(8).SetProp("bondNote","Here!!") d2d = rdMolDraw2D.MolDraw2DSVG(350,300) d2d.drawOptions().setHighlightColour((0.8,0.8,0.8)) d2d.DrawMolecule(cp,highlightAtoms=[],highlightBonds=[19, 8, 9]) d2d.FinishDrawing() SVG(d2d.GetDrawingText())
atom indexはSetAtomMapNumを使っておくと途中でズレたりせず安心だそうです*8。
また、これを使うと追加の描画の設定をしなくてもatom indexが表示されます。
for i, a in enumerate(model_1H.GetAtoms()): a.SetAtomMapNum(i) Draw.MolToImage(model_1H)
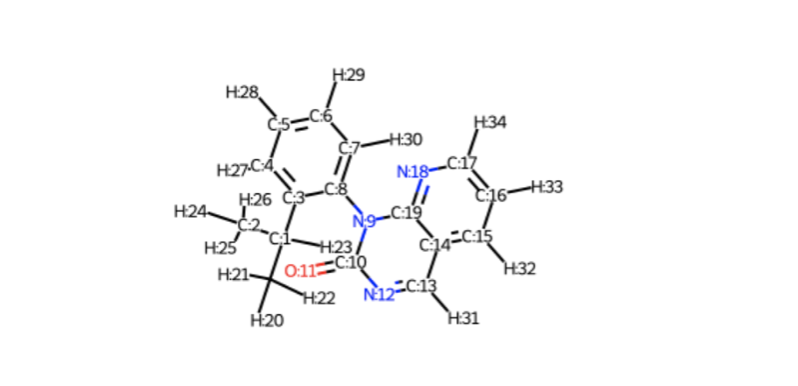
脱線しましたがatom indexがわかりました。
2-2. RDKitの制約付きコンフォマー生成
モデル化合物のMolオブジェクトとatom indexがわかったので、コンフォマーを生成してエネルギー変化を確認します。
目的の2面角に、180°を10°ずつ回転した制約を加えたコンフォマーを作成します。各コンフォマーはMMFF力場で構造最適化します(分子力学法)。
# コード参考元 1; https://future-chem.com/rdkit-constrained-conformer/ # コード参考元 2; https://pschmidtke.github.io/blog/torsion/dihedral/oss/opensource/rdkit/xtb/energy/2021/02/16/torsion-angle-scans-xtb.html # ベースになる3次元構造を生成(再現性のためrandomSeedを設定) AllChem.EmbedMolecule(model_1H,randomSeed=10) # ベースのコンフォマーを取得 conformer=model_1H.GetConformer(0) # ベースが更新されない様にdeepcopyしておく import copy m_1 = copy.deepcopy(model_1H) # MMFF力場の設定 m_1_p = AllChem.MMFFGetMoleculeProperties(m_1) # エネルギーを格納するリスト energy=[] # 回転する角度の範囲 angles=range(0,180,10) # コンフォマーのID = 0からスタート confid=0 # 角度範囲でforループを回す for angle in angles: # MMFF力場 ff = AllChem.MMFFGetMoleculeForceField(m_1, m_1_p) # 目的の2面角に制約を設定(atom indiex : 3, 8, 9, 10) ff.MMFFAddTorsionConstraint(3,8,9,10, False, angle - .1, angle + .1, 10000.0) # 構造の最適化(minimize) ff.Minimize() # エネルギーを取得してリストに追加 energy.append(ff.CalcEnergy()) # 次の新しいコンフォマーを準備 ## 新しいコンフォマーID confid+=1 ## 原子数を引数で指定して新コンフォマーを作成 new_conf = Chem.Conformer(model_1H.GetNumAtoms()) ## 新コンフォマーに原子の位置を設定 for i in range(model_1H.GetNumAtoms()): new_conf.SetAtomPosition(i, (m_1.GetConformer(-1).GetAtomPosition(i))) ## 新コンフォマーにコンフォマーIDを設定 new_conf.SetId(confid) ## 新コンフォマーをベースに追加 model_1H.AddConformer(new_conf)
print(model_1H.GetNumConformers()) # 19
ベースとなるコンフォマーと、0° 〜 170° の10° 刻み18個の合わせて19個が含まれています。アザキナゾリノン骨格をコア構造にしたアラインメントを取って3次元で眺めてみましょう。
# 3次元を眺めるためにpy3Dmolを使う from rdkit.Chem.Draw import IPythonConsole import py3Dmol # 重ねたいコア構造をatomIdで指定してコンフォマーのアラインメントをとる AllChem.AlignMolConformers(model_1H, atomIds=[9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19]) # 3Dビューワーに表示(ベース以外の18個) v = py3Dmol.view(width=600, height=600) for cid in range(1, 19): IPythonConsole.addMolToView(model_1H, confId=cid, view=v) v.setBackgroundColor('0xeeeeee') v.zoomTo() v.show()
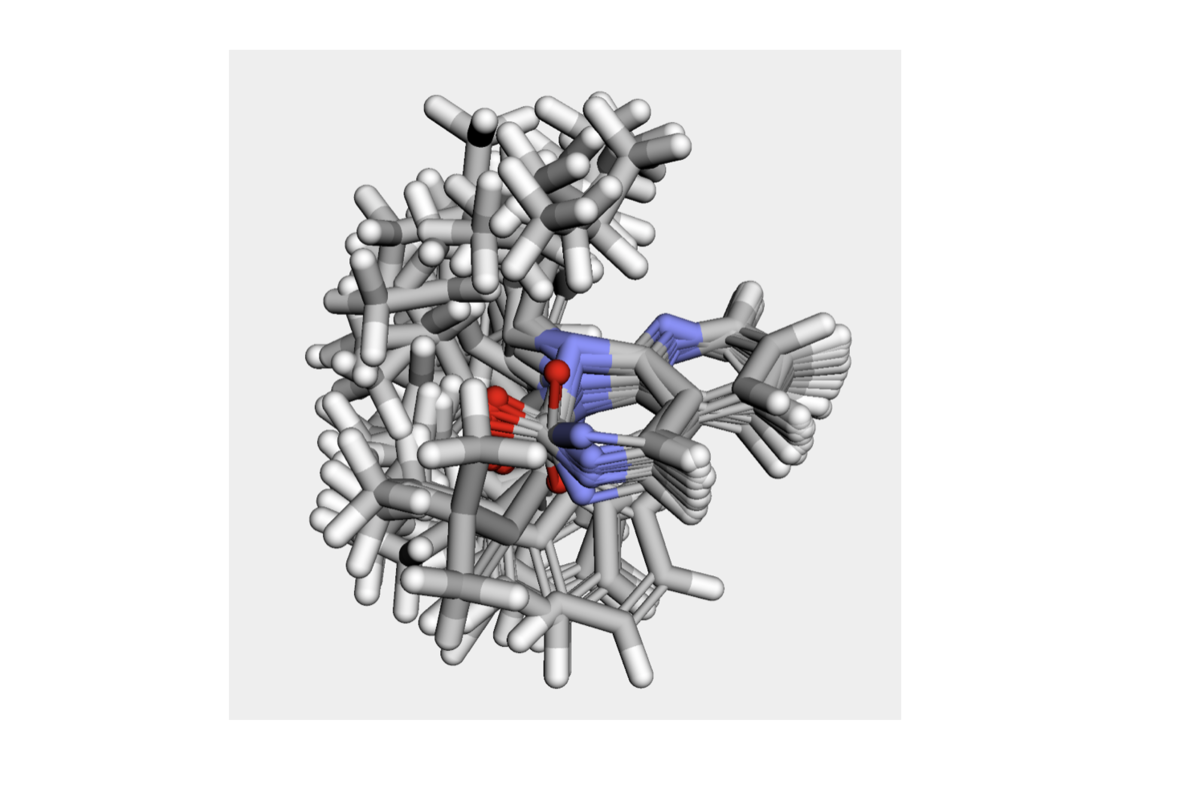
ぐちゃぐちゃですが、とりあえず見たい2面角周りの回転はできていそうです。アザキナゾリノン骨格の平面性がところどころ崩れていることに不安はありますが。。。。
角度とエネルギーの関係をプロットしてみます。
# matplotlibの設定 import matplotlib as mpl import matplotlib.pyplot as plt mpl.style.use('seaborn') # 角度とエネルギーの関係 plt.plot(angles, energy, 'o') plt.xlabel('dihedral angle') plt.ylabel('energy')
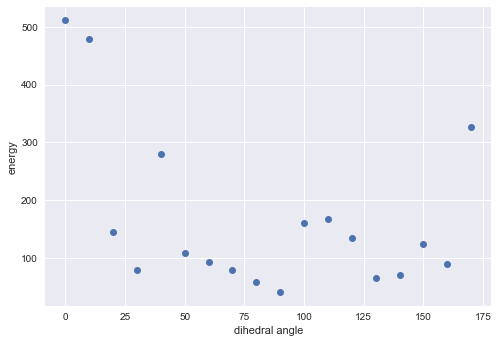
なるほど。よくわからん。。。
2-3. RDKitの制約付きコンフォマー生成~part 2~
先程3Dを眺めた際に骨格の平面性が崩れている感じがありました。どうもこれはRDKitのバージョンによって挙動が違うようです。
参考記事2によるとMMFFのout of plane項の設定に問題があるそうです。記事のおすすめに従ってSetMMFFOopTerm(False)を使ってみましょう。
オプションの設定以外は同じコードです。
# molオブジェクト作り直し model_1 = Chem.MolFromSmiles("CC(C)c1ccccc1n3c(=O)ncc2cccnc23") model_1H = AllChem.AddHs(model_1) AllChem.EmbedMolecule(model_1H,randomSeed=10) conformer=model_1H.GetConformer(0) m_1 = copy.deepcopy(model_1H) # MMFF力場の設定 # さっきとここが違う!Out of plane項の設定を変える。 m_1_p = AllChem.MMFFGetMoleculeProperties(m_1) m_1_p.SetMMFFOopTerm(False) energy=[] angles=range(0,180,10) confid=0 for angle in angles: ff = AllChem.MMFFGetMoleculeForceField(m_1, m_1_p) ff.MMFFAddTorsionConstraint(3,8,9,10, False, angle - .1, angle + .1, 10000.0) ff.Minimize() energy.append(ff.CalcEnergy()) confid+=1 new_conf = Chem.Conformer(model_1H.GetNumAtoms()) for i in range(model_1H.GetNumAtoms()): new_conf.SetAtomPosition(i, (m_1.GetConformer(-1).GetAtomPosition(i))) new_conf.SetId(confid) model_1H.AddConformer(new_conf)
新しく作ったコンフォマーを重ね合わせて眺めてみます。(コードは省略)
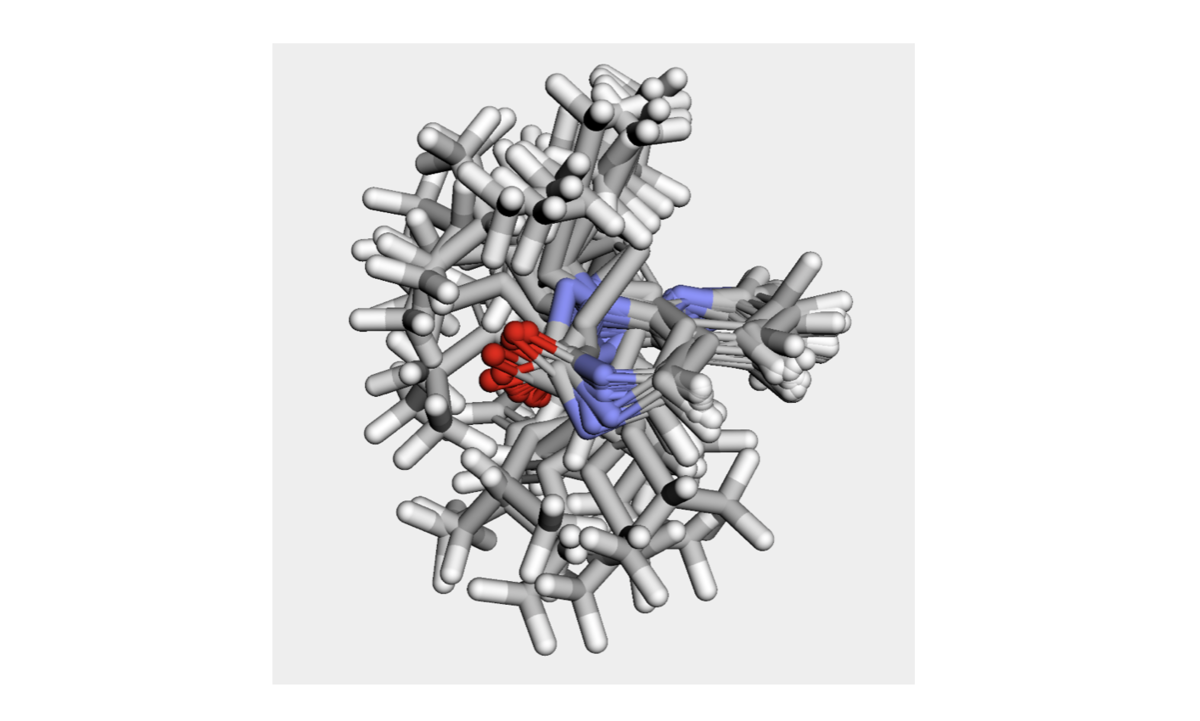
悪化しました。平面性が全然保たれていなさそうです。
一応、こっちでも角度とエネルギーの関係をプロットしてみました。
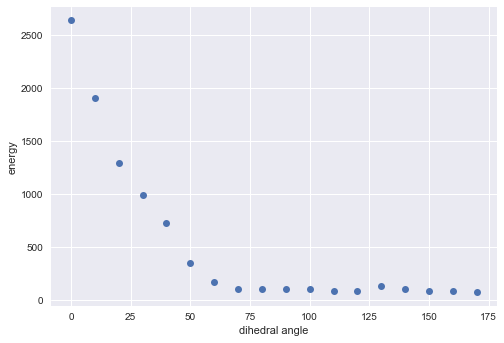
構造が崩れているので参考にはなりませんが、SetMMFFOopTerm()の設定で結果が大きく変わることは分かりました。
2-4. xTBによる2面角スキャン
RDKitのコンフォマー生成を用いた手法がうまくいかなかったので、今度はxTBを使った2面角のスキャンを試したいと思います。こちらも参考記事2で紹介されていたものです。
xTBはオープンソースのSemiempirical Extended Tight-Binding(半経験的拡張強結合近似(?))プログラミングパッケージです。Wikipedia-強結合近似によると、 「系の波動関数を各原子の場所に位置する孤立原子に対する波動関数の重ね合わせにより近似する手法」で「量子化学で用いられるLCAO法と密接な関係がある」そうです。*9
conda-forgeにあるのでcondaでインストールできます。*10
参考記事2で行われている方法は「RDKitで作成したコンフォマーをベースにしてxTBでエネルギー最適化を行おう!」というものです。
ここでは先に発生させたRDKitのコンフォマーを使います(SetMMFFOopTerm(False)していないもの)。インプットが悪すぎるかな?とは思いますが、今回はコードと使い方の学習重視ということでお許しください。
xTBで計算を行うには以下の点に注意すれば良さそうです。
- 入力ファイルの準備: SDFでOK
- 制約をかけたい2面角の原子のインデックスを設定:xTBは1始まりでRDKitの0始まりとズレるのに注意
- 2面角の制約は
.inpファイルに記載する - 出力ファイルは
xtbopt.sdf
各角度の計算結果の構造を保持したいので、参考記事と少しコードを変えています。
# コード参考元 2; https://pschmidtke.github.io/blog/torsion/dihedral/oss/opensource/rdkit/xtb/energy/2021/02/16/torsion-angle-scans-xtb.html # jupyter notebookから実行するためosを利用する import os # 検証したい2面角の角度 angles=range(0, 180, 10) # エネルギーを格納するリスト xtbenergy=[] # 結果の構造を格納するリスト output_structures = [] # rdkitで生成したコンフォマーを使ってループを回して計算 # xTBの入力に使うsdfを作成 for idx,deg in enumerate(angles): w = Chem.SDWriter('mol_1H.sdf') w.write(model_1H,confId=idx+1) w.close() # 2面角のatom indexを設定。1ずれるので(3, 8, 9, 10)ではなく(4, 9, 10, 11) atoms = '4,9,10,11' # 2面角制約の設定に使うxtb input fileを書く fh = open("dihedral_constraint.inp","w") fh.write("""$constrain force constant=1.00 dihedral: {},{} $end""".format(atoms,float(deg))) fh.close() # xTBの実行 # OMP_STACKSIZEは並列計算のための設定(計算環境に合わせて変更) # OMP_NUM_THREADSは計算に使えるスレッドの数(計算環境に合わせて変更) os.system("export OMP_STACKSIZE=4G && export OMP_NUM_THREADS=4,1 && xtb mol_1H.sdf --opt vtight --charge 0 --input dihedral_constraint.inp") # 出力のxtbopt.sdfに含まれる情報からエネルギーを取り出す。 sdr=Chem.SDMolSupplier("xtbopt.sdf") for xtbmol in sdr: xtbenergy.append(xtbmol.GetProp("total energy / Eh")) # 構造を保持する output_structures.append(xtbmol)
結果のエネルギーをプロットしてみます。xtbenergyリストに格納されているのはstr型なのでfloatに変換しておきます。RDKitの計算結果とは単位が異なる(ハートリーエネルギー, Eh)ことにご留意ください。
xtb_energy = [float(i) for i in xtbenergy] plt.plot(angles, xtb_energy, 'o') plt.xlabel('angles') plt.ylabel('xtb_energy')
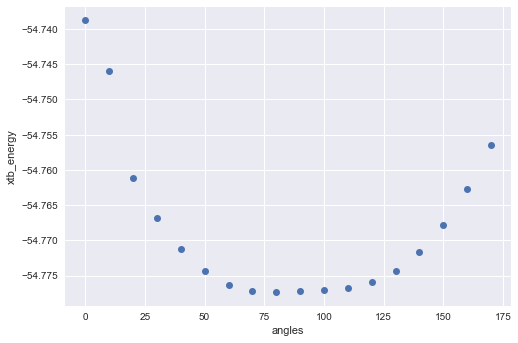
滑らかなプロットになりました。xTBの入力に使ったRDKitの計算結果(一つ目)と見比べると大きな違いです。
構造を眺めてみましょう。
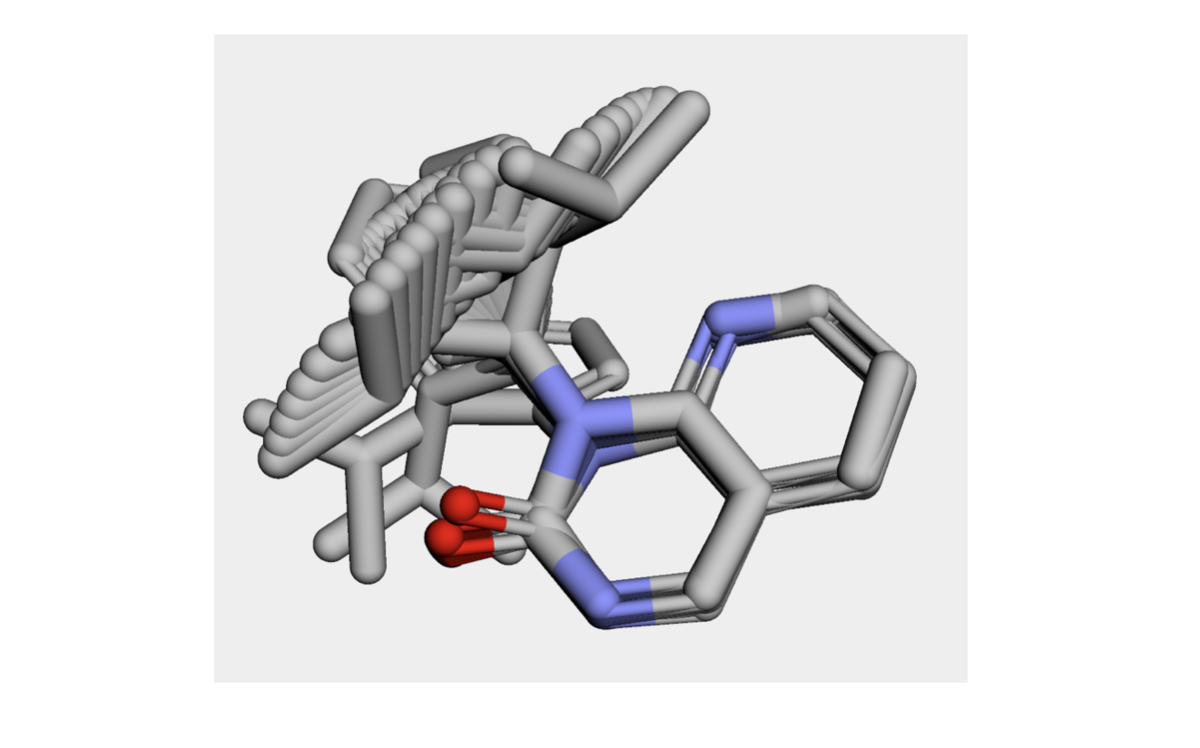
xTBで最適化した構造ではほとんどのコンフォマーで平面性が再現できているようです!!すごい!!計算手法でこんなに変わるんですね!
念のため、最適化に伴って2面角の制約が崩れていないか確認しておきます。
from rdkit.Chem import rdMolTransforms angle_check = [] for m in output_structures: conf = m.GetConformer(0) ang = rdMolTransforms.GetDihedralDeg(conf, 3,8,9,10) angle_check.append(int(ang)) print(angle_check) # [-3, 10, 22, 31, 41, 50, 60, 70, 79, 89, 99, 109, 119, 129, 138, 148, 158, 167]
少々ズレはありますが、10°刻みで0° ~ 170°の18ポイントとる事ができていました。
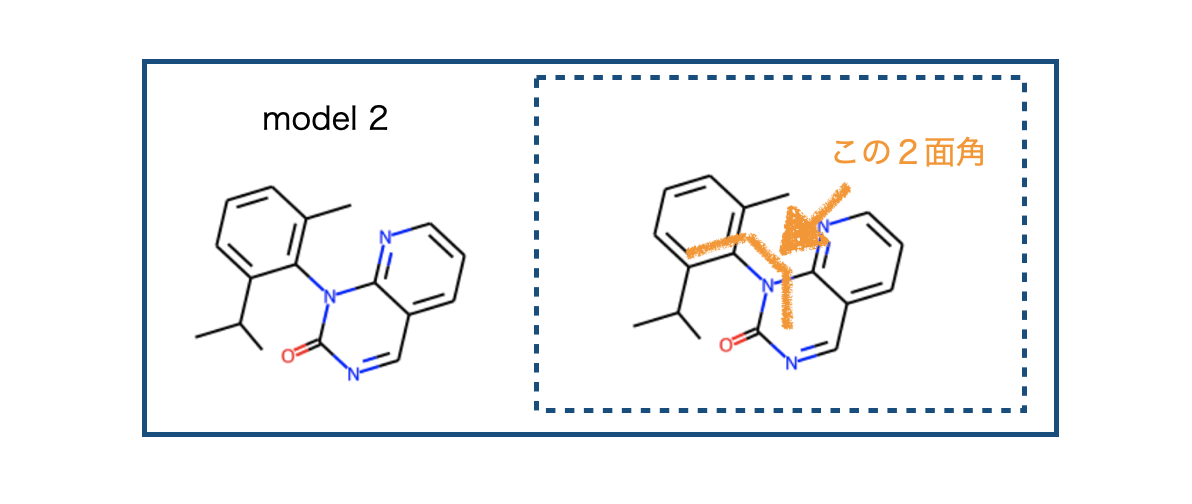
3. 別のモデル化合物2で検証
RDKitのコンフォマー生成とxTBを組み合わせる事でそれらしい結果が得られる事が分かりました。別のモデル化合物で同じ計算を行うとどうなるか、試してみましょう。
# モデル構造作成 model_2 = Chem.MolFromSmiles("CC(C)c1cccc(C)c1n3c(=O)ncc2cccnc23") # 水素原子付加 model_2H = AllChem.AddHs(model_2) # Atom Indexの追加 for i, a in enumerate(model_2H.GetAtoms()): a.SetAtomMapNum(i) Draw.MolToImage(model_2H)
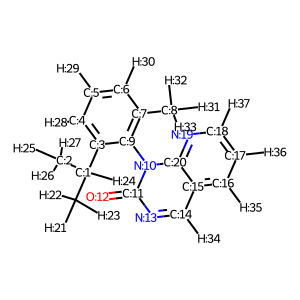
モデル化合物2で目的とする2面角のatom indexは3, 9, 10, 11でした。
xTBに入力するベースのコンフォマーをRDKitで生成します。
# モデル2でコンフォマーを生成 # 初期立体構造の作成 AllChem.EmbedMolecule(model_2H,randomSeed=10) conformer=model_2H.GetConformer(0) m_2 = copy.deepcopy(model_2H) # MMFF力場の設定 m_2_p = AllChem.MMFFGetMoleculeProperties(m_2) energy_2=[] angles=range(0,180,10) confid_2=0 for angle in angles: ff2 = AllChem.MMFFGetMoleculeForceField(m_2, m_2_p) # モデル2に合わせた2面角のatom indexを指定 ff2.MMFFAddTorsionConstraint(3, 9, 10, 11, False, angle - .1, angle + .1, 10000.0) ff2.Minimize() energy_2.append(ff2.CalcEnergy()) confid_2 += 1 new_conf = Chem.Conformer(model_2H.GetNumAtoms()) for i in range(model_2H.GetNumAtoms()): new_conf.SetAtomPosition(i, (m_2.GetConformer(-1).GetAtomPosition(i))) new_conf.SetId(confid_2) model_2H.AddConformer(new_conf)
立体構造とエネルギーはこんな感じでした。
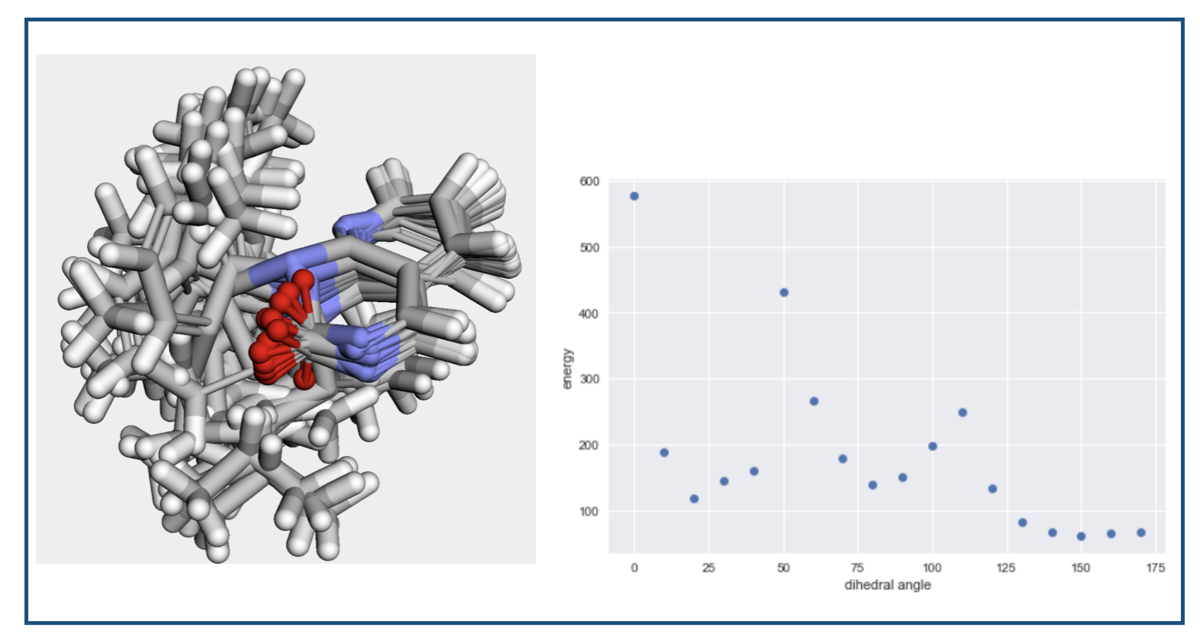
xTBの最適化をおこないます(indexの設定以外は同じです)。
# 検証したい2面角の角度 angles=range(0, 180, 10) # エネルギーを格納するリスト xtbenergy_2 =[] # 結果の構造を格納するリスト output_structures_2 = [] # rdkitで生成したコンフォマーを使ってループを回して計算 # xTBの入力に使うsdfを作成 for idx,deg in enumerate(angles): w = Chem.SDWriter('mol_2H.sdf') w.write(model_2H, confId=idx+1) w.close() # 2面角のatom indexを設定。1ずれるので(3, 9, 10, 11)ではなく(4, 10, 11, 12) atoms = '4, 10,11,12' # 2面角制約の設定に使うxtb input fileを書く fh = open("dihedral_constraint.inp","w") fh.write("""$constrain force constant=1.00 dihedral: {},{} $end""".format(atoms,float(deg))) fh.close() # xTBの実行 # OMP_STACKSIZEは並列計算のための設定(計算環境に合わせて変更してください) # OMP_NUM_THREADSは計算に使えるスレッドの数(計算環境に合わせて変更してください) os.system("export OMP_STACKSIZE=4G && export OMP_NUM_THREADS=4,1 && xtb mol_2H.sdf --opt vtight --charge 0 --input dihedral_constraint.inp") # 出力のxtbopt.sdfに含まれる情報からエネルギーを取り出す。 sdr=Chem.SDMolSupplier("xtbopt.sdf") for xtbmol in sdr: xtbenergy_2.append(xtbmol.GetProp("total energy / Eh")) # 構造を保持する output_structures_2.append(xtbmol)
エネルギーはこんな感じ。
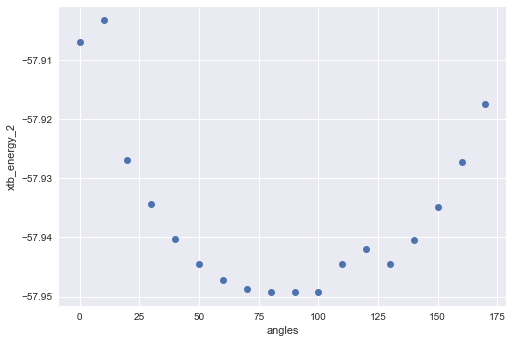
モデル2ではモデル1と比べて置換基を増やしたためか、プロットに谷が2つできました。
立体の確認です。別々のmolオブジェクトとしてリストに格納しているのでAlignMolConformersではなくAlignMolを使っています。
v = py3Dmol.view(width=600, height=600) for mol in output_structures_2: AllChem.AlignMol(prbMol=mol, refMol=model_1H, atomMap=[(i, i) for i in [10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20]]) IPythonConsole.addMolToView(mol, view=v) v.setBackgroundColor('0xeeeeee') v.zoomTo() v.show()
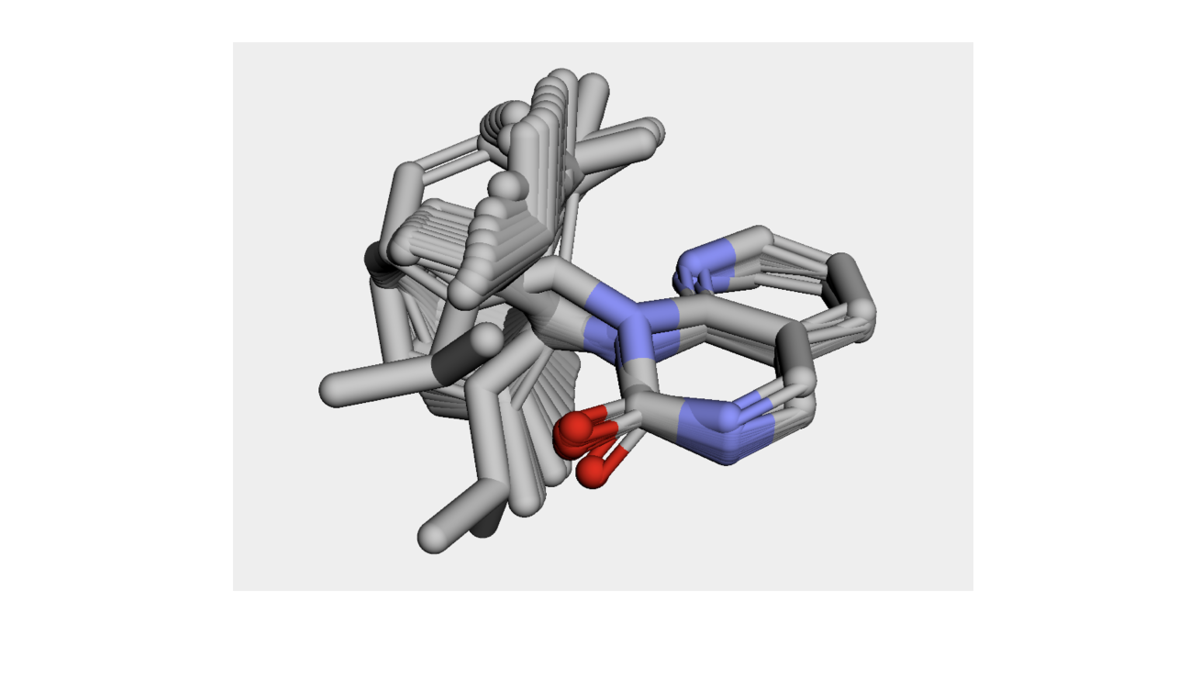
今回もxTBの構造最適化により平面性の崩壊はかなりマシになっているようです。
4. 2つのモデル構造の比較
2つのモデル化合物について2面角とエネルギー変化の計算ができました。合わせてプロットしてみたいと思います。
絶対値での比較はズレが大きいので、それぞれのコンフォマーの最小値との相対的なエネルギー差をとってプロットします。
# それぞれのモデルの最小値と比べた相対的なエネルギーのリスト rel_xtb_energy = [i - min(xtb_energy) for i in xtb_energy] rel_xtb_energy_2 = [i - min(xtb_energy_2) for i in xtb_energy_2] # プロット plt.plot(angles, rel_xtb_energy, label="model_1") plt.plot(angles, rel_xtb_energy_2, label="model_2") plt.xlabel('angles') plt.ylabel('relative_xtb_energy') plt.legend() plt.show
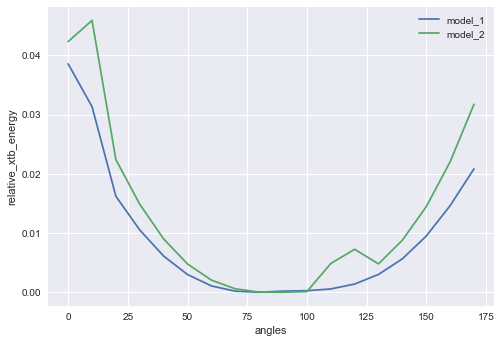
モデル化合物1が青色、モデル化合物2が緑色です。
重ねてみるとどちらも最安定は軸に沿って直交した90°付近にあり、置換基の増えたモデル化合物2は0°、180°付近に近づくに従ってモデル化合物1よりもエネルギーが高く(回転障壁が高く)なっているようです。
それぞれの最大の相対的エネルギー値の差を比較してみましょう。
# 最大の相対的エネルギー値の差 diff_max = max(rel_xtb_energy_2) - max(rel_xtb_energy) print(diff_max, "Eh") # 0.007345450856000468 Eh # 単位を変換(1 hartree = 627.51 kcal/mol) diff_max_kcal = diff_max * 627.51 print(diff_max_kcal, "kcal/mol") # 4.609343866648854 kcal/mol
PC Chem Basics.comさんの記事(1Hartreeは何kcal/mol?(単位換算と物理定数))を基に換算したところ、モデル化合物1と2の最大のエネルギー障壁の差は4.6 kcal/molとなりました。
ところでこれらのモデル化合物はSotorasibの文献中の化合物(18と24)を簡略化したものです。比較するとこんな感じ。
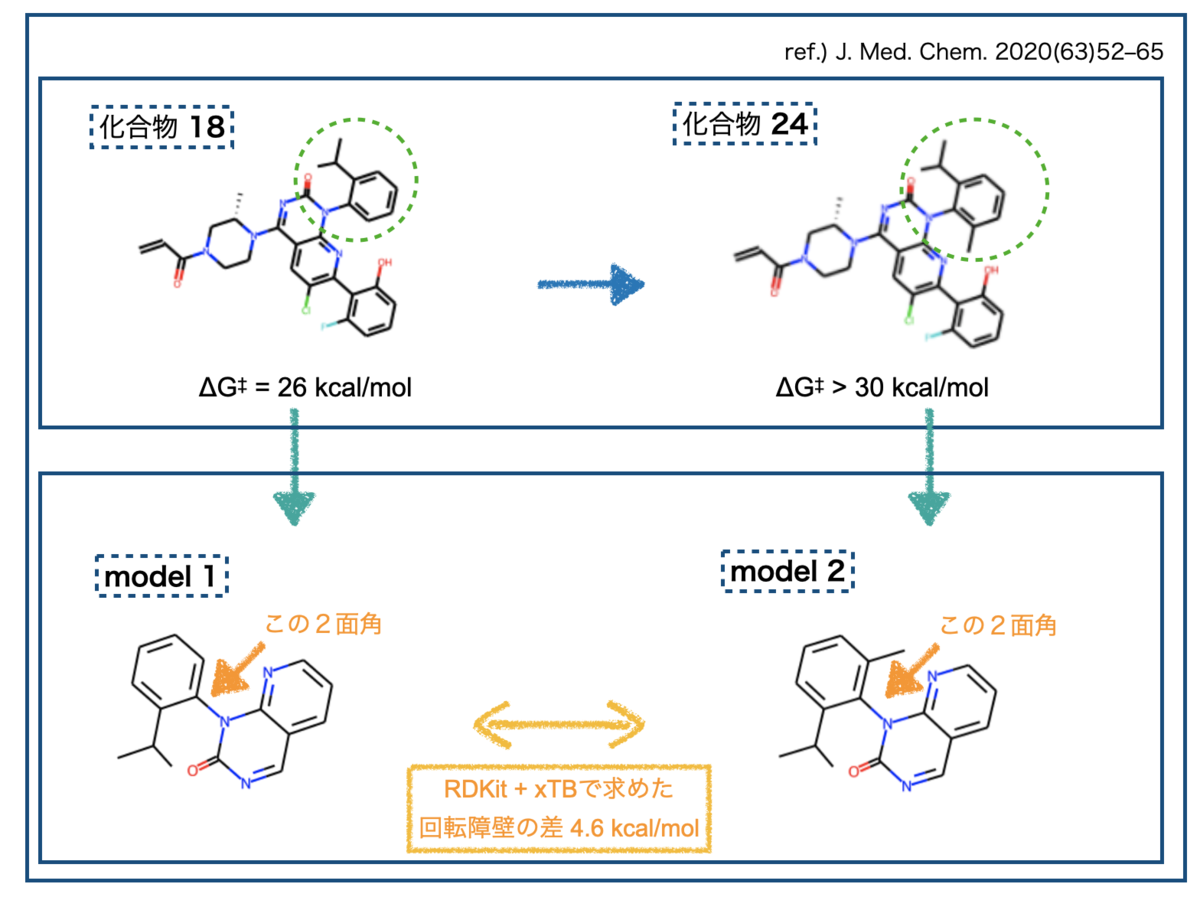
化合物18から24への変換でエネルギー障壁は4 kcal/mol以上上昇したとのことですが、今回のモデル化合物の検証結果は4.6 kcal/molでした。
ひょっとして結構良い結果でてる????
おわりに
以上、今回は医薬品と回転異性体をテーマに、その実例の紹介と回転障壁を題材とした計算を試してみました。
回転異性体は構造式をパッとみてわかる不斉中心が無いので分かりづらいように思います(私だけ??)。こんな時、気軽にちょっと計算してみてエネルギー差がわかったりしたら「あーはいはいそんな可能性もあるのねー」と納得しやすく便利な気がします。
計算の題材としては、近年の話題の医薬品Kras G12C阻害薬Sotorasib(AMG510)文献上の化合物をベースにしてみました。
インターネットの賢い人々の検討結果をコピー&ペーストしたところ、意外にも(?)それらしい結果が得られて正直びっくりでした。素晴らしい情報を公開してくださっている皆様に感謝です。
実際には色々な問題点に目をつぶった、無理矢理な議論をしてしまっています。xTBでマシになったとはいえ平面性が崩れたりしていました。。。
もっと良い方法があるよ!お前のやり方はあまりにもひどい!頭悪すぎ!等々、ご意見いただけると嬉しいです。
なお、本記事はtorsion driveって用語格好いい!と思って書きました。*11
Calculation drives new chemistry!!!
知らんけど
*1:言わずもがな、こちらもノーベル化学賞(2001年)ですね
*2:アレン(allene)みたいなそもそも回転できないものもあります。念のため。
*3:参考: FutureMed.Chem.(2018)10(4),409–422
PuMedでFull textよめます。
*4:もう一つACSのwebinar資料を参考にしています。こちらは何故かインターネット上でPDFが閲覧できるものの出どころ不明なので引用は避けます。
*5:Creative Commonsでは無さそうなので図表の引用は控えます
*6:図中の構造式は論文のSupporting Informationで提供されているSMILESをもとにRDKitで描画しました。
*7:他にはこちらの記事のようにatom indexを確認する方法もあるそうです:RDKitで原子に番号を表示できなくて困っていた話
*8:参考:【RDKit】RWMol で分子(Mol instance) を編集する
*9:誰か説明してください
*10:mambaが使えるならそっちの方が速いかも…
*11:創薬アドベントカレンダーの@iwatobipen先生の記事 Compare torsion drive results between C and O linker of specific structure を読んで、回転って面白いなと思ったのがきっかけです。アドカレに間に合わなくてすみません。
DeepMindのDFTのやつ(DM21)がColabで動きました
こんな感じでDM21がColab上で動かせました。コードはDM21のGitHubのREADMEから抜粋しました。
メタンの計算で14分かかりました。何が計算されたのかいまいち分かっていないです。
1. DM21のインストール
!pip install git+git://github.com/deepmind/deepmind-research.git#subdirectory=density_functional_approximation_dm21
2. 必要なライブラリのインポート
DM21はPySCFというソフトウェアに基づいて作られているので基本的にはPySCFと同じやり方で計算できるそうです。残念ながら私はPySCFを使った事がありません。
import density_functional_approximation_dm21 as dm21 from pyscf import gto from pyscf import dft
3. 計算したい分子の用意
メタン分子を構築するための情報はDM21のGitHubのREADMEを利用しています
# Create methane methane = gto.Mole() methane.atom = """H 0.000000000000 0.000000000000 0.000000000000 C 0.000000000000 0.000000000000 1.087900000000 H 1.025681956337 0.000000000000 1.450533333333 H -0.512840978169 0.888266630391 1.450533333333 H -0.512840978169 -0.888266630391 1.450533333333""" methane.basis = 'def2-qzvp' methane.verbose = 4 methane.build()
分子が構築できるとこんな結果がでます。
[CONFIG] conf_file None
[INPUT] verbose = 4
[INPUT] num. atoms = 5
[INPUT] num. electrons = 10
[INPUT] charge = 0
[INPUT] spin (= nelec alpha-beta = 2S) = 0
[INPUT] symmetry False subgroup None
[INPUT] Mole.unit = angstrom
[INPUT] 1 H 0.000000000000 0.000000000000 0.000000000000 AA 0.000000000000 0.000000000000 0.000000000000 Bohr
[INPUT] 2 C 0.000000000000 0.000000000000 1.087900000000 AA 0.000000000000 0.000000000000 2.055833050914 Bohr
[INPUT] 3 H 1.025681956337 0.000000000000 1.450533333333 AA 1.938257988385 0.000000000000 2.741110734552 Bohr
[INPUT] 4 H -0.512840978169 0.888266630391 1.450533333333 AA -0.969128994193 1.678580657029 2.741110734552 Bohr
[INPUT] 5 H -0.512840978169 -0.888266630391 1.450533333333 AA -0.969128994193 -1.678580657029 2.741110734552 Bohr
nuclear repulsion = 13.4613239153198
number of shells = 57
number of NR pGTOs = 209
number of NR cGTOs = 177
basis = def2-qzvp
ecp = {}
CPU time: 6.55
DFT計算するためのソルバーを作成
RKSはRestricted Kohn-Shamの略の様です。DM21汎関数をソルバーに代入することで、DM21を使った計算ができる様になります。
# Create a DFT solver and insert the DM21 functional into the solver.
mf = dft.RKS(methane)
mf._numint = dm21.NeuralNumInt(dm21.Functional.DM21)
DFT計算の実行
%%time
# Run the DFT calculation.
mf.kernel()
******** <class 'pyscf.dft.rks.RKS'> ********
method = RKS-RHF
initial guess = minao
damping factor = 0
level_shift factor = 0
DIIS = <class 'pyscf.scf.diis.CDIIS'>
diis_start_cycle = 1
diis_space = 8
SCF conv_tol = 1e-09
SCF conv_tol_grad = None
SCF max_cycles = 50
direct_scf = True
direct_scf_tol = 1e-13
chkfile to save SCF result = /content/tmpmc1ww10e
max_memory 4000 MB (current use 1094 MB)
XC library pyscf.dft.libxc version 5.1.7
S. Lehtola, C. Steigemann, M. J. Oliveira, and M. A. Marques, SoftwareX 7, 1 (2018)
XC functionals = LDA,VWN
P. A. M. Dirac, Math. Proc. Cambridge Philos. Soc. 26, 376 (1930)
F. Bloch, Z. Phys. 57, 545 (1929)
S. H. Vosko, L. Wilk, and M. Nusair, Can. J. Phys. 58, 1200 (1980)
radial grids:
Treutler-Ahlrichs [JCP 102, 346 (1995); DOI:10.1063/1.469408] (M4) radial grids
becke partition: Becke, JCP 88, 2547 (1988); DOI:10.1063/1.454033
pruning grids: <function nwchem_prune at 0x7fce59db0710>
grids dens level: 3
symmetrized grids: False
atomic radii adjust function: <function treutler_atomic_radii_adjust at 0x7fce59db0b90>
small_rho_cutoff = 1e-07
Set gradient conv threshold to 3.16228e-05
tot grids = 53998
関係なさそうな部分を省略します。SCFサイクルが走ってる感じの記述がずっと並んでました。
最終的な結果は以下
Extra cycle E= -40.5178537368252 delta_E= 4.76e-10 |g|= 5.27e-08 |ddm|= 6.94e-08
converged SCF energy = -40.5178537368252
CPU times: user 25min 26s, sys: 12.7 s, total: 25min 38s
Wall time: 13min 41s
収束したSCFエネルギーは「 -40.5178537368252」(単位はHartree?)で、計算に14分弱かかりました。
GitHubの計算結果は「'CH4': -40.51785372584538」だったのでほとんど同じ値が出ていそうです。
思ったより計算時間がかかりました。
DeepMindのDFT論文を読んだメモ
今年はDeepMindの論文祭りなのでしょうか?数学者とのコラボレーションがNatureに発表されたかと思いきや、今度はScienceにDFTに関する文献が出されたそうです。
数学ははなから諦めていますし、理論化学もさっぱりですが、やっぱりDFTの方はちょっと知りたい。
というわけでDFT論文、大体こんな話ですかー?っていう感想文です。

- 1. Science文献が公開されてた!
- 2. 前置き
- 3. 論文の中身
- 4. どうしてDeep Learningがよかったんだろう?
- 5. DFT x Deep Learningって他にもみたことあるけど?
- 6. おわりに
1. Science文献が公開されてた!
Scienceの文献自体はオープンアクセスではありませんが第一著者のJames Kirkpatrick博士がResearchgateにアップロードしてくださっています。
下記、DeepMindのブログからもリンクされているので正当な手続きで公開されているはず。。。*1
またOpen AccessのPDFがこちらから閲覧できます。こっちも上の記事からリンクされてました。
2. 前置き
2-1. 前置き① ~ DFTの未解決問題? ~
そもそもDFTにおける未解決問題とは何だったのでしょうか?
多分こういう話です。
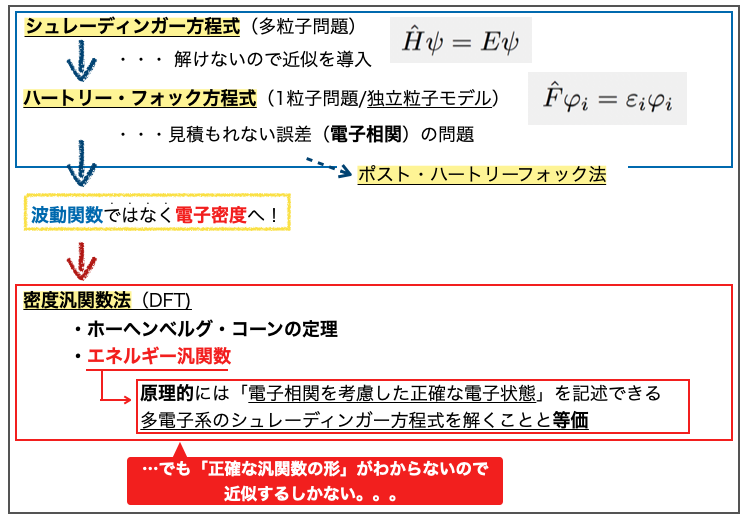
シュレーディンガー方程式はそのままでは解けないので近似の導入によりハートリー・フォック方程式を解く問題に変換されました。 それでも見積もれない誤差(電子相関 の問題)があるため様々なポスト・ハートリーフォック法が出てきました。
これに対して波動関数ではなく電子密度から出発しても系の状態(物理量)を知ることができるよ!そっち解こうぜ!ってのが密度汎関数法(DFT)のアプローチです。
ポスト・ハートリーフォック法ではさまざまな電子間の相互作用を考慮するため、電子数が多くなる(系が大きくなる)と計算量が膨大になり現実的に解くのが難しくなってしまうそうです。DFTは「より計算量が少なくて済むため現実的な時間で解くことができる」という利点がある様です。
ちょっとDFTに関する専門用語を貼ってそれっぽくします。

電子密度から系の状態がわかるというのはホーヘンベルグ・コーン定理に基づくそうです。この定理から電子密度とエネルギーの関係性を表すエネルギー汎関数というものがでてきます。
DFTではエネルギー汎関数に基づき原理的には「電子相関を考慮した正確な電子状態」を記述することができ、多電子系のシュレーディンガー方程式を解くのと等価になるそうです。
ここで問題となるのが「エネルギー汎関数の正確な形がわからない」ということで、これがDFTの未解決問題にあたるようです。
2-2. 前置き② ~ コーン・シャム近似と交換相関汎関数 ~
さて、エネルギー汎関数は正確な形がわからないため様々な近似モデルが提案されました。 初期のモデルでは精度が悪くて非実用的だったようですが、より改善したコーン・シャム近似が導入されたことでDFTが実用的なものになったそうです。
ざっとこんな感じみたいです。

コーン・シャム近似では、それ以前の近似モデルとは異なる新しい項、交換相関汎関数(exchange-correlation functional)が追加されています。
以前のモデルの精度の悪さは運動エネルギー汎関数の近似の悪さと考え、運動エネルギーを正確・計算可能な形に表したうえで、表現仕切れない残りを交換相関汎関数に押し込めています。また、交換相関汎関数には、電子間相互作用エネルギーのうち電子間クーロンエネルギーで近似できない残りも押し込められています。
よくわかりませんが、うまく表せない部分を押し込めて煮詰めた闇鍋部分が交換相関汎関数ってことですね(???)
3. 論文の中身
3-1. DeepMindのDFTモデル(DM21)
また前置きが長くなってしまいました。本題のDeepMindの文献についてです。
この論文ではDM21と名付けたDeepLearningに基づく汎関数(functional)が提案されています。
DFTでは「電子密度とエネルギーとの間に関係がある」ことがわかっていますが、その「エネルギー汎関数の正確な形がわからない」というのが大きな問題でした。
「未解決のブラックボックスな関数がある。。。それならデータから学習した最高精度のモデルで置き換えちゃえばいいじゃん!」って話ですね!・・・たぶん。
より具体的には標準的なコーン・シャム近似(Kohn-Sham DFT)のうち、交換相関項(exchange-correlation term)を学習させて置き換えています。先に見た闇鍋部分ですね!
学習の概要は論文のFig. 1をご参照いただくとして、大体こんな感じです。
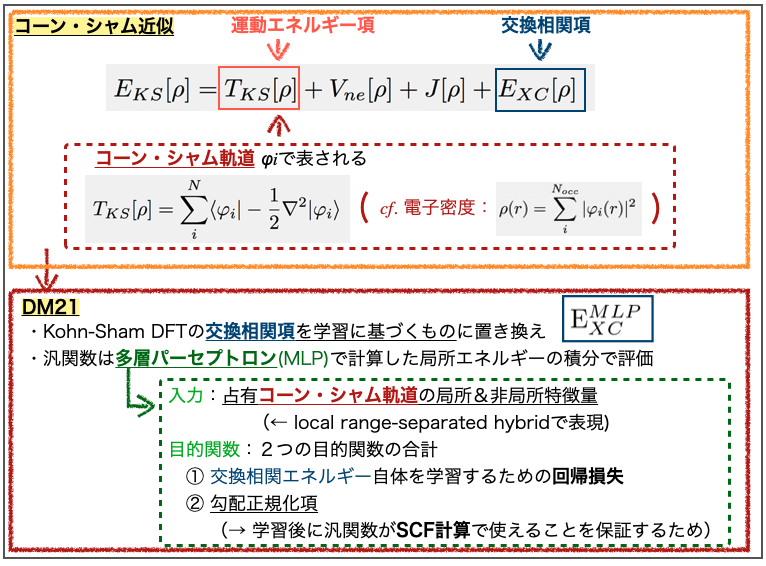
多層パーセプトロン(mulitlayer perceptron, MLP)の入力は占有されたコーン・シャム軌道(occupied Kohn-Sham orbitals)の局所的(local)特徴量と非局所的(nonlocal)特徴量です。コーン・シャム軌道はコーン・シャム近似で運動エネルギー項を表すために導入された波動関数に類似した形のものです。
この入力は「local range-separated hybrid」で表現できるそうですが、これが何かよくわかりませんでした。局所的な距離(r1, r2,…, rG)で区切ったものの組み合わせ(hybrid)という意味でしょうか??? お分かりの方ご教示いただけると嬉しいです。
また、学習の目的関数としては2つの目的関数の合計を用いています。ひとつは交換相関エネルギー自体を学習するための回帰損失(regression loss)で、もう一つは勾配正規化項(gradient regularization term)です。
前者はわかりやすいですが、後者は学習後の推論フェーズで重要になるようです。勾配正規化項を目的関数としておくことで、汎関数の導関数(derivative)が自己無撞着場の手続き(self-consistent filed (SCF) calculation)で利用できることが保証されるそうです。
DFTの計算ではコーン・シャム方程式を解くことが行われますが、この方程式は行列形式に置き換えられ固有値を求める問題となります。この固有値問題をとくための繰り返しの手続きがSCF計算と呼ばれるもののようです。
3-2. DM21が改善した2つの課題
さて、DeepMindの提唱するDFTモデル、DM21で行われていることがぼんやり見えてきました。ではDM21でどの様な問題が解消されたのか?より具体的にみてみます。
何より名は体を表す!
論文のタイトル「Pushing the frontiers of density functionals by solving the fractional electron problem」が表す通り「fractional electronの問題」です!・・・何のこっちゃ???
DeepMindのブログ記事によると、既存のDFT計算には長年解決できなかった2つの課題、①非局所化の誤り(the delocalization error)と②スピン対称性の破れ(spin symmetry breaking)があったそうです。
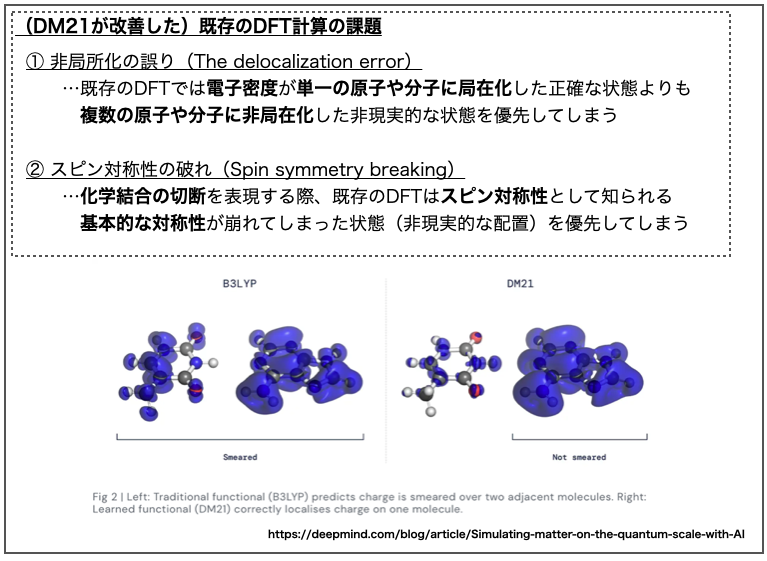
要するにDFTの既存の汎関数の近似モデルは不完全なので、「物理化学的に考えたら現実的にはこうだよね!」ってのと違う計算結果になってしまう、ってことみたいです。電子が1個、2個ではなく、分数個(fractional)あるとか言われても気持ち悪いですよねー。
ってことで、それぞれもう少し見てみます。
3-2-1. 非局在化の誤り
「① 非局所化の誤り」についてはDeepMindのブログ記事の図(Fig. 2)が分かりやすいので上に引用させていただきました。*2
上図ではDNAの塩基ペア(アデニン(A)-チミン(T))間における電荷の分布が計算されています。左が従来の手法(B3LYP)、右がDeepMindの手法(DM21)で計算した結果で、ブルーの網がけで電荷密度(charge density)が表現されています。
DNAには遺伝情報を伝えるという役割以外にも、電荷を輸送する(charge transport)という物理化学的にとても面白い性質が報告されています。2011年にはNature Chemistryにこんな論文も出ています。
この性質を理論的に理解しようとするとDFTの出番!となります。DNAの構成要素である塩基ペアの部位で「イオン化した状態で電荷がどの様に分布しているか?」知りたい、というモチベーションです。
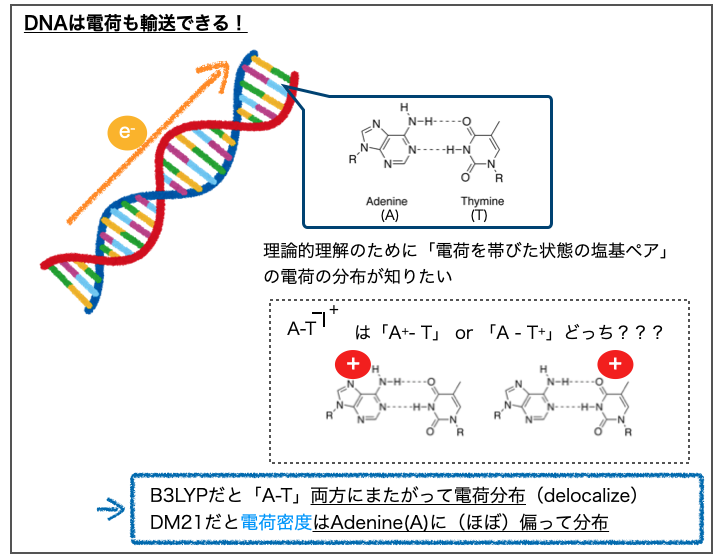
A-Tペアについてこの計算を行った結果が先に引用したFig. 2です。
アデニンがプラス電荷を帯びた「A+-T」の方が「A-T+」よりも安定というのが正解の様ですが、B3LYPでは(アデニンへの偏りはあるものの)A,T両方にまたがって電荷が分布しています。一方でDM21では(ほぼ)アデニンのみに電荷密度が偏った結果が得られています。
DFTにおいて「非局在化した計算結果が得られやすい」という課題が改善されていることの良いデモンストレーションになってますね。
3-2-3. スピン対称性の破れ
もう一方のDFTの課題「②スピン対称性の破れ」はそのままの話で「基本的には対称に配置されている方が安定のはずのスピンについて、対称性が崩れた結果をより安定と評価してしまう」ということの様です。
私では論文であげられている例を理解できなかったので、皆様Fig. 3B,3Cをご参照のうえ、ご教示いただけると嬉しいです。
なんかこんな感じの話。

もうひとつあげられている例はビシクロブタンの異性化メカニズムの遷移状態についてでした。(こっちもよくわからなかった)
3-3. DM21のモデルの中身をもう少し ~ fractional chargeとfractional spin ~
DM21が解決しようとしたDFTの課題についても雰囲気がつかめてきました。では「何故、DM21のモデルでうまくいったのか?」。成功理由と関係のありそうなモデルのデザインの中身をもう少しみてみます。
DM21の取り組んだ課題は大きくは「fractional electronの問題」でした。この問題に対するため、DM21はfractional electronを有する系についての2つのタイプの制約条件、①fractional charge(FC)と②fractional spin(FS)に従う様にデザインされたそうです。
FCとFSはそれぞれ「非整数の総電荷(noninteger total charge)の系」と「非整数スピン磁性化(noninteger spin magnetization)の系」です。これらの系は当然、非現実的な架空のもの(fictitious)です。
ですが、「現実の電荷密度もFCやFSの特徴を持つ領域を含みうる」ので、この「理想状態の問題を正確にモデル化」することができれば、「多様な分子や素材についても上手く機能する汎関数」が得られるのではないか?という考えに基づくようです。
現実には無い架空の特性を持つユニットをモデルにくみこむあたりは、AlphaFold2でアミノ酸残基をガスとしてモデル化してフィッティングしていた解き方に通じるところがあって面白いです。
さて、fractional electronの問題を解消するために使われたFCとFSですが、これらの制約条件はデータで表現する事ができるため、これに従って分子系のエネルギーを再現する様に汎関数を学習すれば良い、ということになるらしく、Deep Learningと相性の良い問題に帰着できたことになったようです。
手法とモデル化がうまく合致していてすごい!
3-4. fractional chargeとfractional spinをもう少し
架空のモデル、fractional charge (FC) とfractional spin (FS) ですが、これらはDM21で初めて導入されたものではなく、既存のDFT汎関数でも使われていました。ただし、従来の汎関数ではFCとFSに関連したエラーが解消できず問題となっていた様です。
このエラーは①電荷を帯びた分子と、②閉殻中性分子(closed-shell neutral molecules)において結合の切断の表現が不正確になってしまう、という問題です。
詳細は論文のFig. 2をご参照いただくとして、大体こんな話です。

それぞれ、①の問題はDFTの「電子密度の非局在化」の課題、②はDFTの「スピン対称性の破れ」の課題と関連していることが何となく分かりますね。
これらのFCとFSに関する問題について、DM21は従来の汎関数を上回るパフォーマンスが得られたので、先にみた実課題での検証(A-T, hydrogen chain, etc.)に進んだ、というのが論文の流れです。*3
以上が、DeepMindのデザインしたDFT汎関数DM21の概要と、その解決しようとした課題についてでした。
論文についてはここまでで、あとは雑感です。
4. どうしてDeep Learningがよかったんだろう?
さて、今回もDeepMindはDeep Learningで分野の壁を突き破ったわけですが、何故DFTの課題にDeep Learningを適用するのがよかったのでしょうか?
今回の論文を受けたNatureのNews記事(DeepMind AI tackles one of chemistry’s most valuable techniques)から引用します。
“It’s sort of the ideal problem for machine learning: you know the answer, but not the formula you want to apply,” says Aron Cohen, a theoretical chemist who has long worked on DFT and who is now at DeepMind. (Nature 600, 371 (2021))
「答えがわかっていて、わかっていない式の形を求めるのは機械学習にとって理想的な問題」だそうです。
この考えの背景にあるのはおそらく「普遍近似定理(universal approximation theorem)」ではないでしょうか?

上記は2層についての定理ですが、さらに多層にすることの利点は層の深さに応じて表現力が増す、適応力があがるといったことにある様です。
詳細は上記の書籍や、東京大学大学院 鈴木大慈先生がSlideShareに公開してくださっている資料(深層学習の数理)などをご参照ください。
雑な理解ですが、多層のニューラルネットワークであれば「任意の関数を精度よく近似」できるので、「関数の形がわかっていない問題」にDeep Learningを適応していい感じにすれば「良い近似モデルが得られる!」ってことになりそうです。
5. DFT x Deep Learningって他にもみたことあるけど?
最後に何故この研究にインパクトがあったのか?ちょっと想像してみます。
というのも「DFTに対してDeep Learningを適用して改善した!」という論文・ニュース記事などは偶に目にするので何が違うのかなー?と気になったからです。
例えばこういうのです。

どちらもニューラル・ネットワーク・ポテンシャル(NNP)の研究です。
一つ目はフロリダ大学A.E. Roitberg教授らのグループによるもの(Chem. Sci., 2017,8, 3192-3203)で、2つめはPreferred Computational Chemistry社の提供するプラットフォームMatlantisに関する論文です(arxiv:2106.14583)。
私自身が専門分野外のため、これらの論文を比較対象として引用するのは間違っているかもしれません。ごめんなさい。
DFT計算はシュレーディンガー方程式を扱うよりも計算コストが低いという利点がありますが、それでも系が大きくなるにつれて必要な計算量が増え、非常に時間とコストのかかるようになってしまうようです。「計算のシミュレーションに時間かかるなら、実際に実験した方が速くない??」って言われそうです。
この課題があるため、Deep Learningで同等の計算結果が出せる様に学習し、計算時間のかかる部分を置き換えることで加速・コストを下げよう!というのがNNPの取り組みの様です。
何となくですが、これらの研究は計算コストの改善に焦点があり、既存のDFTが抱える精度上の課題の解決とはなっていなさそうです(たぶん)。
DM21は汎関数自体を作って、根本的な課題に取り組んだ、と言う点にインパクトがあったのかな?と思います。
6. おわりに
以上、今回はDeepMindのDFTに関するScience文献を読んでみた感想文でした。
量子化学に疎く、DFTの計算もやった事がなかったのでそもそも既存の汎関数に課題があることも知りませんでした。というより、論文を読み終えた今でもいまいち課題を理解しきれていなかったりします。
DFTの課題でスピン対称性の破れが上がっていたのに、Fractional spinのところではスピン非局在化の過剰評価が問題になっていて、結局対称性が破れるのと破れないのどっちが問題なの???となってしまいました。私に量子化学は難しすぎます。
なんとなくですが、適切なデータがある問題であれば、Deep Learningでうまく関係性を記述するモデルを作る事ができそうだ、ということが分かったのはよかったです。DeepMindが次にどんな課題に取り組むのか?楽しみですね!
ところで今回の論文のDM21ですが、素晴らしいことにGitHubで公開されています。ぜひぜひ専門家の皆様の使ってみた感想がお聞きしたいところです。
なお、今回の記事の前置き部分などは以前に書いたブログ記事を使いまわしています。
ずっと雰囲気から成長できていなくてすみません。今回も色々と間違いが多そうなのでご指摘いただけると嬉しいです。
ではでは!
アラート ≠ 禁止 〜ZINCデータベースの医薬品にPAINSフィルターをかけてみた話〜
創薬(dry)アドベントカレンダーを埋めてみます。アドベントカレンダーの記事を見ると「いよいよ年末かー」となりますね。
さて、今年も色々な格好いい分子が出てきてケミカルスペースは広大だなぁと思いました。
というわけで発表します!びっくりモルキュールオブザイヤー!
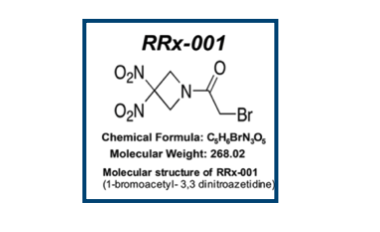
抗がん剤として臨床開発中だそうです。論文は以下。
- Discovery of RRx-001, a Myc and CD47 Downregulating Small Molecule with Tumor Targeted Cytotoxicity and Healthy Tissue Cytoprotective Properties in Clinical Development
J. Med. Chem. 2021(64)7261
ニトロ基2つにアミドのα位にハロゲン原子。。。見ただけで涙がちょちょ切れそうです。
これを臨床開発するなんて本当にびっくりですが、逆にいうと常識(?)みたいなものに囚われて有用性が見過ごされている分子がまだまだあるかもしれません。
そこで今回は「承認済みの医薬品の中にどれくらい忌避構造(アラート構造)が含まれているか?」調べてみることにしました。
1. この記事でやること
やることは単純です。ZINC15データベースにあるFDA approved drugsの一覧を使って、RDKitでPAINSフィルターにかかる分子がどれくらいあるか探してみるだけです。
2. データの準備
「ZINCデータベースは、バーチャルスクリーニングのために特別に準備された、市販の化学物質に関する精選されたコレクション」だそうです(Wikipedia - ZINCデータベースより)。
カリフォルニア大学サンフランシスコ校(UCSF)のJohn J. Irwin博士の研究室とBrian K. Schoichet博士の研究室により開発・維持されているそうで、現在はZINC20です。
- ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery
J. Chem. Inf. Model 2020(60)6065
サーバーが混んでいてアクセスできなかったので今回はZINC 15を使います。
- ZINC 15 – Ligand Discovery for Everyone
J. Chem. Inf. Model 2015(55)2324)
よく知らないけどよく見るからこれを使っとけば良いんでしょう!知らんけど。
こんなページです。
ここにSubstancesがあるじゃろ。これをこうして。こうじゃ。
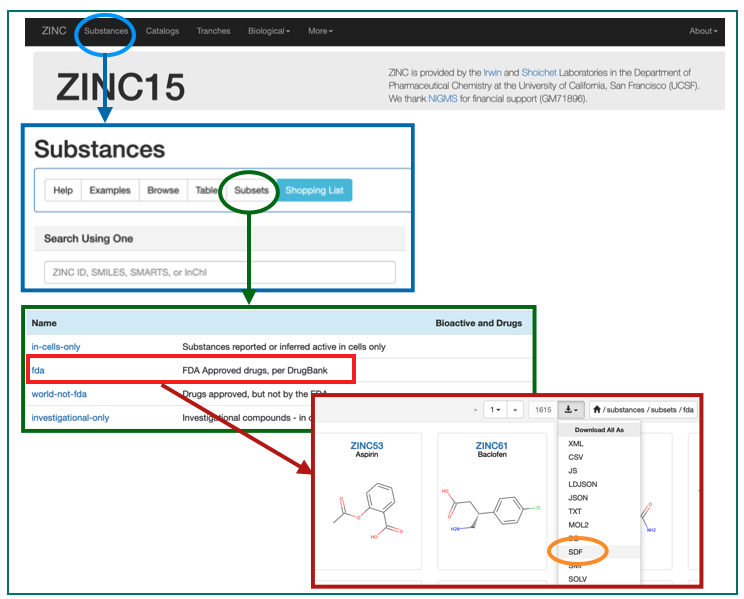
これでFDA承認済みの医薬品構造のSDFが手に入りました!
2015年に公開されたデータベースのようなので、少し古いかもしれませんが今回はこれで。。。
3. PAINS
3-1. PAINSフィルターって?
PAINSフィルターについては「化学の新しいカタチ」さんのブログ(RDKitのPAINSフィルターで化合物をスクリーニング)でわかりやすく解説してくださっています(RDKitのコードも!) 。
PAINSはPan-Assay INterference compoundsの略だそうです。
- New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays
J. Med. Chem. 2010(53)2719
ある種の部分構造を持つ化合物は、ハイスループットスクリーニング(HTS)を実施した際に、色々なアッセイ(pan-assay)で活性化合物としてヒットしてくるそうです。このような化合物は偽陽性である可能性が高くなるため要注意です。
そんなアッセイの妨害になる化合物(interference compounds)をフィルタリングして取り除けるように、「予め要注意な部分構造をリスト化しておこう!」というわけで作られたのがPAINSフィルターみたいです。
3-2. PAINSフィルターをもう少し
PAINSフィルターの具体的な中身についてもう少し見てみましょう!
2018年 ACS Chemical BiologyにPAINS公開後7年を経て再度その内容を振り返るといった趣旨の文献が出されています。こちらはオープンアクセスとなっています*2
- Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations
ACS Chem. Biol. 2018(13)36
「PAINSは数秒で簡単に100個も1000個も大量に化合物選別できて便利だけどブラックボックスのまま使うのはやめてね!」ってな感じです。
そもそもPAINSに上げられている構造はどうしてアッセイの邪魔になってしまうでしょうか? 文献では以下の様な特徴が指摘されています。

タンパク質や試薬と反応してしまう構造(求電子剤、光反応剤)は非特異的な不可逆反応を起こしそうなのでわかりやすいですが、キレート剤やフォトクロミズムといったものは考えてもみませんでした。確かにアッセイの原理と相性が悪そうです。なるほど!
さてPAINSフィルターをブラックボックスとしないために把握しておくべき注意点ですが、PAINSが「商用化合物を使ってPPI(protein-protein interaction)阻害を対象に行われた6つのHTS(AlphaScreen)における観察」に基づくものであることに由来する様です。*3
注意点はこんな感じ。

PAINSフィルターを使うときは用法・用量を守って適切に!
参考までに②のそもそもライブラリに含まれていなかった構造の例としてFig. 1を引用しておきます。

ついでにFig. 2も面白いので引用しておきます。メカニズムも用語も色々あってわかりにくいから整理して分類したよ!って図です。
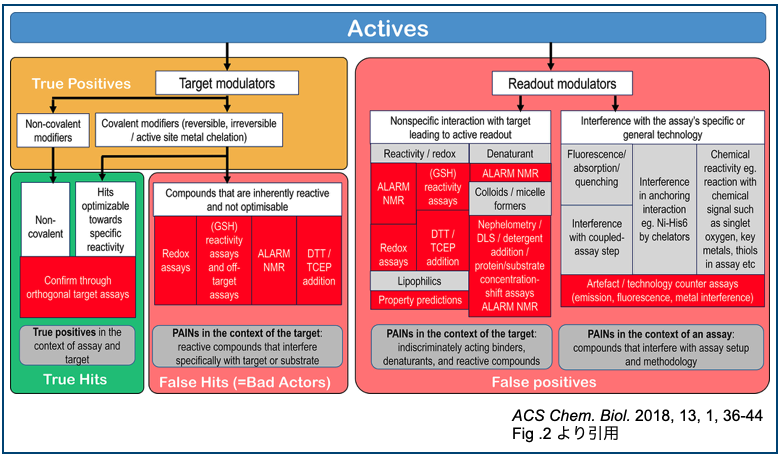
promiscuous binderとかfrequent hitter、bad actorってさらっと言えたら業界人っぽくないですか?・・・そうでもないですか。すみません。
4. RDKitのPAINSフィルターをつかってみよう
4-1. 簡単な使い方 ~ RRx-001を例に ~
だいたいPAINSのやりたいことがわかったので早速使ってみましょう!
まずは記事冒頭にあげたRRx-001を例にRDKitのPAINSフィルターを使ってみます。コードは化学の新しいカタチさんの記事とRDKitのブログ記事を参考にさせていただいています。
とりあえず化合物構造を準備します。
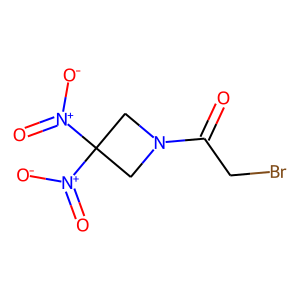
from rdkit import Chem from rdkit.Chem import AllChem, Draw # PubChemよりRRx-001のSMILESを取得(PubChem ID : 15950826) RRx_001_smiles = "C1C(CN1C(=O)CBr)([N+](=O)[O-])[N+](=O)[O-]" # Molオブジェクトを作って描画 RRx_001_mol = Chem.MolFromSmiles(RRx_001_smiles) Draw.MolToImage(RRx_001_mol)
RDKitのPAINSフィルターには4つ(PAINS、PAINS_A、PAINS_B、PAINS_C)あります。ABCはそれぞれPAINSを作る際のHTSで150以上の化合物に含まれた構造(Family_Filter_A)、15 - 149化合物(Family_Filter_B)、14以下(Family_Filter_B)に対応しているようです。
よくわからないので「PAINS(無印)」を使ってみす。
FilterCatalogに実装されており、FilterCatalogParamsオブジェクトに使いたいフィルタリング要素を追加してフィルターを作った後、目的の化合物に適用すれば良いそうです。
# インポート from rdkit.Chem import FilterCatalog # パラメータのオブジェクトを作る param = FilterCatalog.FilterCatalogParams() # PAINSフィルターをパラメータに追加する param.AddCatalog(FilterCatalog.FilterCatalogParams.FilterCatalogs.PAINS) # フィルターを作成 filt = FilterCatalog.FilterCatalog(param)
フィルターができたので適用します。
HasMatchで「フィルター中の部分構造が含まれているかどうか?」を確認することができます。
print(filt.HasMatch(RRx_001_mol)) # False
False !!! RRx-001はPAINSフィルターにはかからない化合物でした!見た感じヤバいのに。。。
他のフィルターではどうでしょうか?RDKitには他に3つのフィルター(BRENK、NIH、ZINC)が用意されています。それぞれ適用してみましょう。
# フィルター名のリスト filter_names = ["BRENK", "NIH", "ZINC"] # 順番に適用 for name in filter_names: p = FilterCatalog.FilterCatalogParams() fc = "FilterCatalog.FilterCatalogParams.FilterCatalogs." + name p.AddCatalog(eval(fc)) f = FilterCatalog.FilterCatalog(p) res = f.HasMatch(RRx_001_mol) print("Does filter " + name + " detect RRx-001? : " + str(res) ) # Does filter BRENK detect RRx-001? : True # Does filter NIH detect RRx-001? : True # Does filter ZINC detect RRx-001? : False
BRENKとNIHではRRx-001はフィルターにかかりました!どの構造が認識されたのでしょうか?
GetMatchesで「具体的にどのようなフィルターにひっかかったか?」取り出せるそうです。NIHを例に行ってみます。
# NIHフィルターを作成 p2 = FilterCatalog.FilterCatalogParams() p2.AddCatalog(FilterCatalog.FilterCatalogParams.FilterCatalogs.NIH) f2 = FilterCatalog.FilterCatalog(p2) # フィルターに認識された構造情報を取り出す matches = f2.GetMatches(RRx_001_mol) for match in matches: print(match.GetProp("description")) print(match.GetProp("Reference")) print(match.GetProp("Scope")) print("------------") """ alpha_halo_carbonyl Jadhav A, et al. Quantitative Analyses of Aggregation, Autofluorescence, and Reactivity Artifacts in a Screen for Inhibitors of a Thiol Protease. J Med Chem 53 (2009) 37D51. doi:10.1021/jm901070c. annotate compounds with problematic functional groups ------------ non_ring_ketal Jadhav A, et al. Quantitative Analyses of Aggregation, Autofluorescence, and Reactivity Artifacts in a Screen for Inhibitors of a Thiol Protease. J Med Chem 53 (2009) 37D51. doi:10.1021/jm901070c. annotate compounds with problematic functional groups ------------ primary_halide_sulfate Jadhav A, et al. Quantitative Analyses of Aggregation, Autofluorescence, and Reactivity Artifacts in a Screen for Inhibitors of a Thiol Protease. J Med Chem 53 (2009) 37D51. doi:10.1021/jm901070c. annotate compounds with problematic functional groups ------------ """
「alpha_halo_carbonyl、non_ring_ketal、primary_halide_sulfate」の3つの部分構造が認識されること、NIHフィルターが以下の文献をもとにしていることがわかりました。
- Quantitative Analyses of Aggregation, Autofluorescence, and Reactivity Artifacts in a Screen for Inhibitors of a Thiol Protease
J. Med. Chem. 2010 (53)37
このままではわかりにくいので構造式上で確認してみましょう。フィルターにかかった構造のアトム番号はGetFilterMatchesで取り出せるそうです。
# descriptionのリスト descriptions = [match.GetProp("description") for match in matches] # アトム番号のリストのリスト atom_nums_list = [] for match in matches: atom_nums = [x[1] for x in match.GetFilterMatches(RRx_001_mol)[0].atomPairs] atom_nums_list.append(atom_nums) # 描画 Draw.MolsToGridImage([RRx_001_mol for _ in range(len(matches))], highlightAtomLists=atom_nums_list, legends=descriptions)
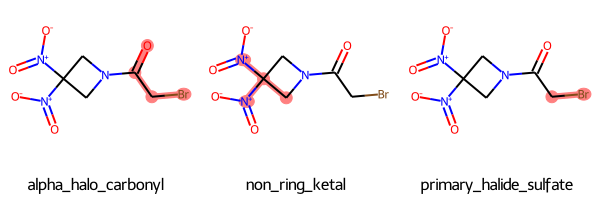
構造を描画するとわかりやすいですね!臭素原子はカルボニルα位のハロゲンや、primary-halideとして認識されています。意外にも(?)、ニトロ基はニトロ基としてではなく、同一炭素原子に2つの窒素原子が結合した構造(non_ring_ketal)として認識されている様です。
同じことの繰り返しなので省略しますがBRENKフィルターでは以下の結果が得られました。
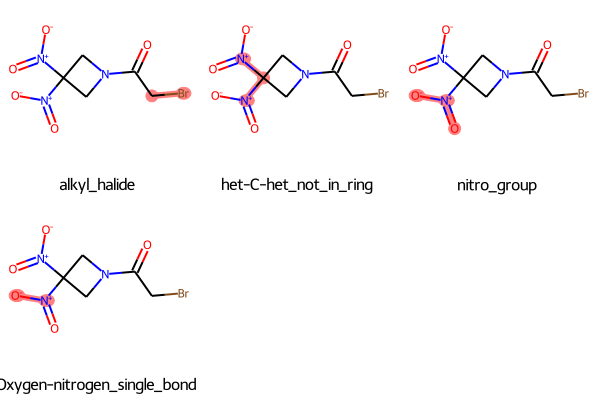
今度はニトロ基はnitro_group、Oxygen-nitrogen_single_bondとしても認識されている様です。見比べると面白いですね!
4-2. いざ承認化合物に適用!
RDKitのPAINSフィルターの使い方が大体わかりました。
それではようやく本題!ZINCデータベースから取り出したFDA承認化合物に適用してみましょう!
フィルターとしてはPAINSを使い、それぞれの化合物に「フィルターにマッチする部分構造がいくつ含まれているか?」検証してみます。
化合物数が多いのでPandasToolsでDataFrameとして扱います。
from rdkit.Chem import PandasTools import pandas as pd # SDFをデータフレームに読み込む df = PandasTools.LoadSDF("fda.sdf") print(df.shape) # (1615, 4) df.head(2)
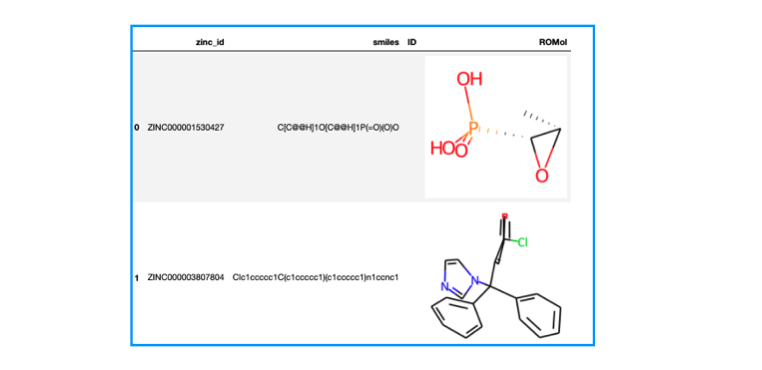
1615個の化合物がありました。zinc_id、smilesといったプロパティが含まれており、化合物は3次元に起こされた座標が入っている様です。
PAINSフィルターとマッチする構造を数える関数を作ってmap()でDataFrameに適用します。
# PAINSフィルター作り直し param = FilterCatalog.FilterCatalogParams() param.AddCatalog(FilterCatalog.FilterCatalogParams.FilterCatalogs.PAINS) filt = FilterCatalog.FilterCatalog(param) # フィルターにかかった数を数える関数 def match_num(mol): if filt.HasMatch(mol): matches = filt.GetMatches(mol) num = len(matches) else: num = 0 return num # PAINSフィルターにかかる化合物か否か?(True or False) df['PAINS'] = df.ROMol.map(filt.HasMatch) # フィルターにかかった回数は? df['filter_match_num'] = df.ROMol.map(match_num)
そもそもFDA承認化合物にPAINSフィルターにかかったものはあったのでしょうか?
# PAINSフィルターでTrueになった化合物の数 PAINS_drugs_num = df['PAINS'].sum() print("PAINS detected {} compounds.".format(PAINS_drugs_num)) # PAINS detected 79 compounds.
# 割合 PAINS_ratio = round((PAINS_drugs_num / len(df))*100) print("ratio : {} %".format(PAINS_ratio)) #ratio : 5 %
79化合物がPAINSフィルターにひっかかることがわかりました!
承認済み1615化合物の約5% にあたります。結構多いですね。
フィルターにかかった回数はどうでしょうか?
print(df['filter_match_num'].value_counts()) """ 0 1536 1 72 2 5 3 2 Name: filter_match_num, dtype: int64 """
72化合物で1回、5化合物で2回、2化合物で3回ひっかかっているようです。
3回かかった2化合物を取り出してみましょう。
df[df['filter_match_num'] == 3]
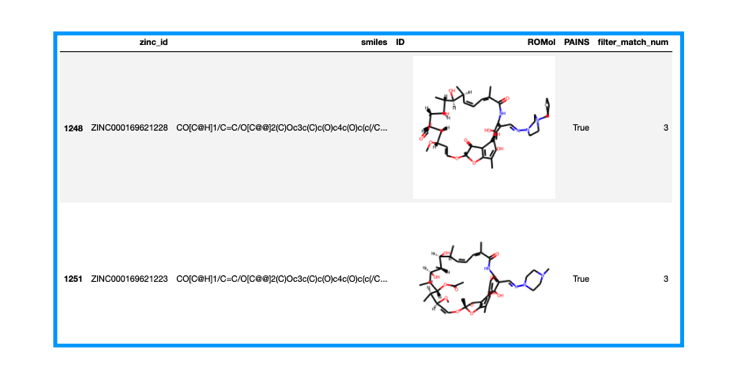
3次元だとよくわからないので2次元に描画しなおしてみます。マクロサイクルの化合物みたいなのでSetPreferCoordGenで描画の調整を行います。
# 該当化合物のSMILESを取り出してMolオブジェクトを作りなおす df3 = df[df['filter_match_num'] == 3] mols = [Chem.MolFromSmiles(smi) for smi in df3["smiles"]] # ZINC idを取り出す zinc_ids = [zinc_id for zinc_id in df3["zinc_id"] ] # マクロサイクルの描画を綺麗にする設定 Draw.rdDepictor.SetPreferCoordGen(True) # 描画 Draw.MolsToGridImage(mols, legends = zinc_ids)
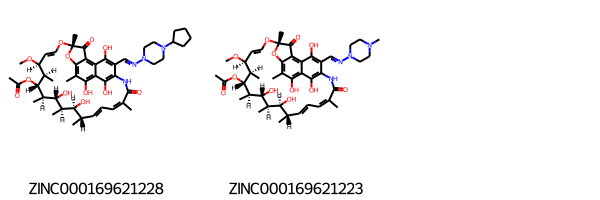
PubChemで調べたところ、それぞれCyclopentylrifampicinとRifampicineのようです。
大体同じ構造なのでRifampicineでPAINSフィルターに合致した構造をみてみます。コードは繰り返しなので省略します。
最初の二つはヒドラゾン構造とその周辺が認識されている様です。もう一つはケトンと共役したナフトール構造でした。
二重結合と共役したアミド構造やビニルエーテルなど他にも気になる構造はありますがこれらは認識されていない様ですね。
5. おわりに
以上、今回は見た目のインパクトが強いRRx-001を題材にRDKitのPAINSフィルターの使い方をお勉強し、ついでにZINC15から取得したFDA承認済み化合物にも適用してみました。
途中で引用したACS Chemical Biology「PAINS振り返り論文」中に、「PAINSは網羅的なものではない」という注意点が指摘されていました。実際に、RRx-001はPAINSには認識されないものの、他のフィルター(BRENK、NIH)ではアラートとして認識されていました。
また、FDA承認済み化合物とPAINSの関係についても上記文献に以下の記載がありました。
A small proportion (ca. 5%) of FDA-approved drugs contain PAINS-recognized substructures, these comprising both natural products and synthetic drugs.
ZINC15の例でも確かに5% でフィルターに引っかかりました。中でも3回フィルターにひっかかった化合物はRifampicineとCyclopentylrifampicin(リファペンチン)でした。
この2つは抗結核薬で、放線菌から取り出されたリファマイシンをリード化合物としたリファマイシン系化合物に分類されるもののようです。確かにいかにも天然物由来な構造です。
天然物由来の化合物には部分構造だけを取り出してみたら「そんな構造大丈夫なの?」と不安になるものが他にも多そうです。人間がデザインしたライブラリには絶対に入らなさそうな構造に有用性が見出されるのが天然物の面白さですよね!
先入観にとらわれなければライブラリにも自然界にもまだまだお宝が眠っているかもしれません。フィルターは適切に使いたいですね!
ところで、昨年に引き続き今年も対面で人に会うのが難しい一年でした。
来年は会えん 痛みがなくなるといいですね!
おしまい!
化学構造認識(OCSR) DECIMERプロジェクトとDECIMER 1.0で遊んだ話
前回の記事で化学構造式のOCRソフトImg2Molで遊んでみました。「画像に含まれる化学構造式を認識してSMILESに変換」してくれるというものでした。
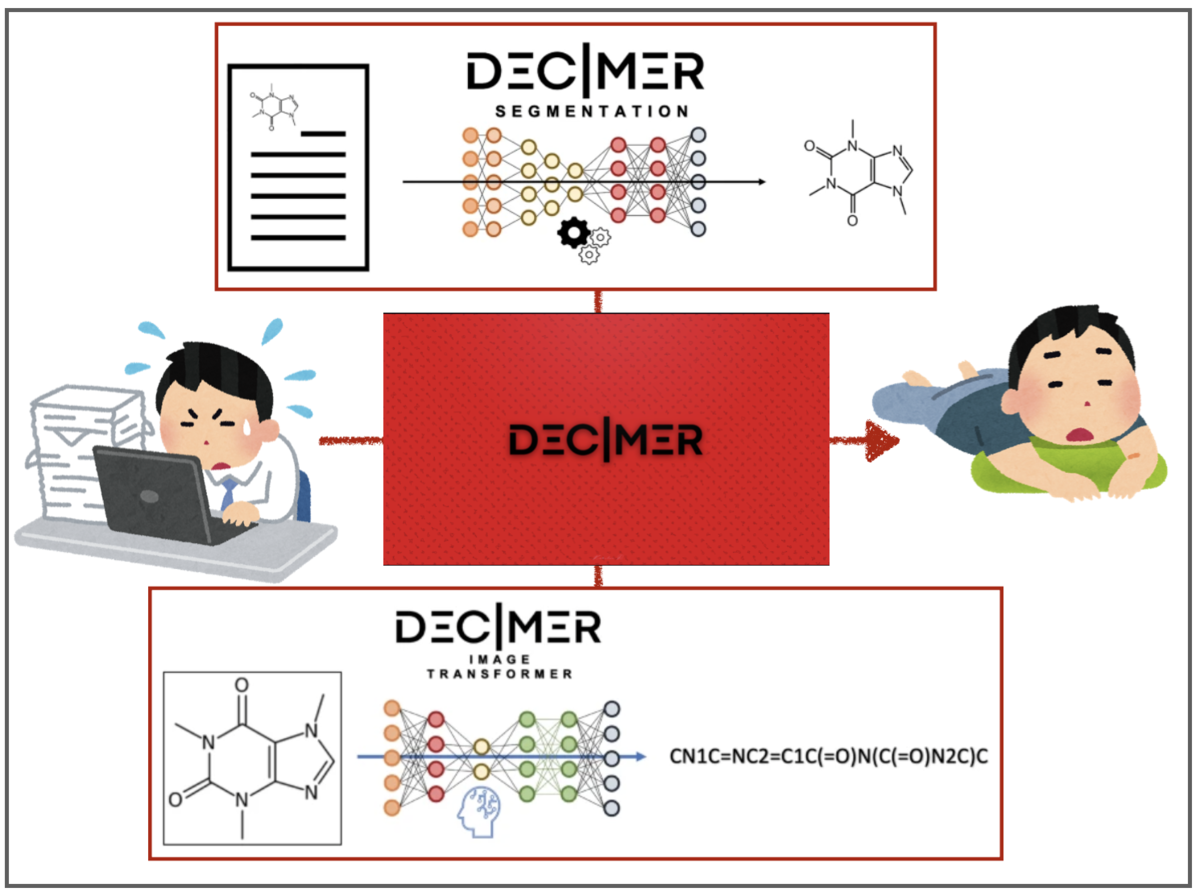
Img2Molと同時期に同様のタスクを扱うソフト DECIMER 1.0が公開されています。
- 文献①(オープンアクセス) : DECIMER 1.0: deep learning for chemical image recognition using transformers J. Cheminform. 2021(13)61
- GitHub : DECIMAR-Image_Transformer
こちらもコードや学習済みパラメータを公開してくださっています‼︎ Img2Molとの比較も兼ねて遊んでみましょう!
1. DECIMER文献の流し読み
1-1. DECIMERプロジェクトについて
さて、Img2Molは製薬会社バイエルの機械学習チームによるものでしたが、DECIMERはドイツのFriedrich-Schiller大学Chirstoph Steinbeck教授の研究室(HPはこちら)で開発されたアカデミックな研究に由来するものです。
こちらもディープラーニングによる手法で、DECIMERはDeep lEarnig for Chemical ImagE Recognitionの略です。
このソフトは「化学構造式の光学認識(Optical Chemical Structure Recognition, OCSR)」の課題について、「最新の人工知能技術を使ったオープンソースの自動化されたソフトウェアを開発しよう!」というDECIMERプロジェクトの中の一つです(プロジェクトWebサイト)。
関連する文献として、今回取り上げる文献の前のバージョン(DECIMER(無印))や、、、
- 文献②:DECIMER: towards deep learning for chemical image recognition J. Cheminform. 2020(12)65
テキストと画像の入り混じった文献を区分けして、画像部分をとりだすセグメンテーションのためのツール、、、
- 文献③:DECIMER-Segmentation: Automated extraction of chemical structure depictions from scientific literature J. Cheminform. 2021(13)20
があります(どちらもオープンアクセス)。後者のツールはウェブアプリとして利用可能になっています。
全て筆頭著者はKohulan Rajanさんとなっていますので、こちらの方が中心になって進められているようですね。すごい。
1-2. DECIMER 1.0のモデル
それではDECIMER 1.0の具体的な中身を見てみましょう!Img2Molと同じくディープラーニングに基づく手法ですが、具体的な中身は結構違います。
おさらいですが、Img2Molは画像を畳み込みニューラルネットワーク(CNN)で認識し、化学構造式の特徴を表現する空間(CDDD)に一度埋め込んだあと、線形表記(Canonical SMILES)として取り出す、という2段階の構成でした。またCDDDはオートエンコーダーによるものでした。
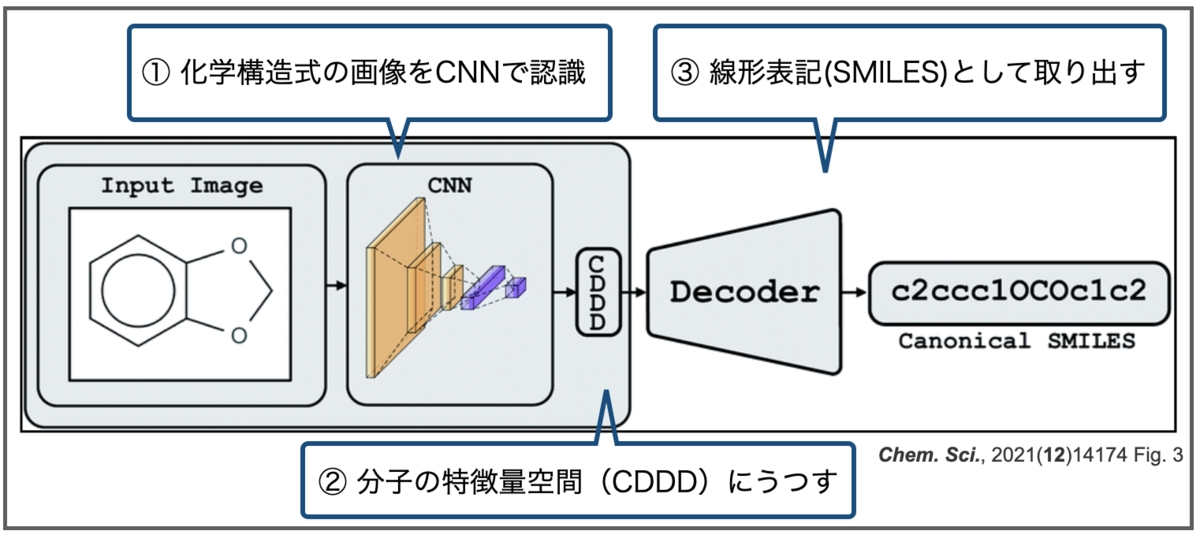
対してDECIMER 1.0も、画像を認識するための入力ネットワークと、認識した情報から化学構造式を取り出すためのネットワークの2段階の構成となっています。
ですが、化学構造を取り扱うネットワークを構築するために線形表記 SELFIESも利用していること、またTransformerベースのモデルになっていること、といった異なる点もあるようです。
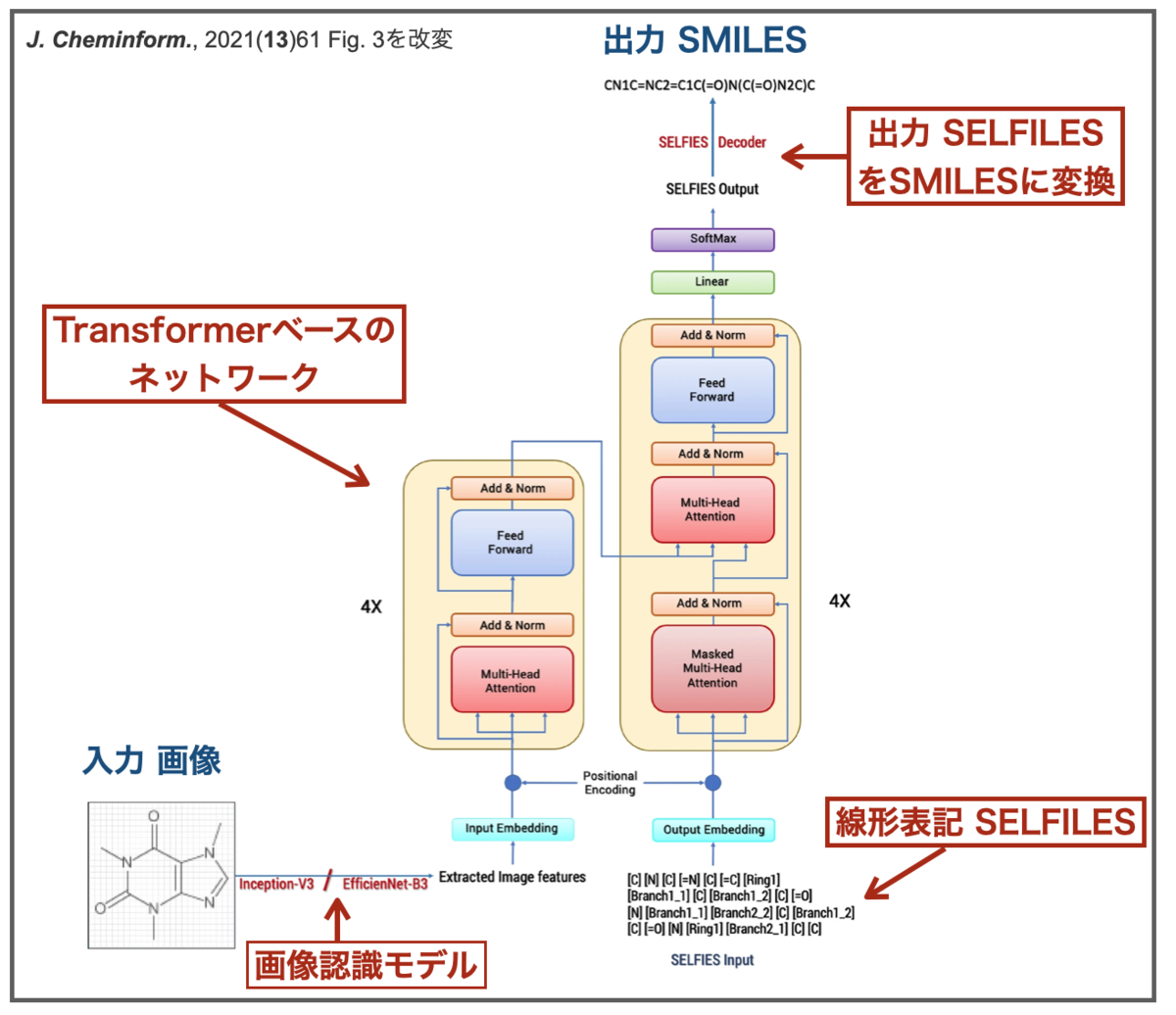
SELFIES (Self-referencing embedded strings)はAspuru-Guzik教授らのチームが開発した新しい化合物の線形表記方法で、この文法に従えばどんなふうに書いても100%分子として意味をなすという特徴があるそうです。
以下の記事が日本語でわかりやすく解説してくださっているのでおすすめです。
また、モデルのベースとなっているTransformerですが、こちらはAlphaFold2のベースにもなっていたやつですね。自然言語処理の分野で高い性能を発揮したらしく、よく使われているらしいです。
なお、前報DECIER(無印)ではオートエンコーダーをベースとしたモデルが使われています。DECIMER 1.0でもencoder-decoder型のモデルとTransformer型モデルが比較されており、結果、後者の方が精度が良かったそうです。
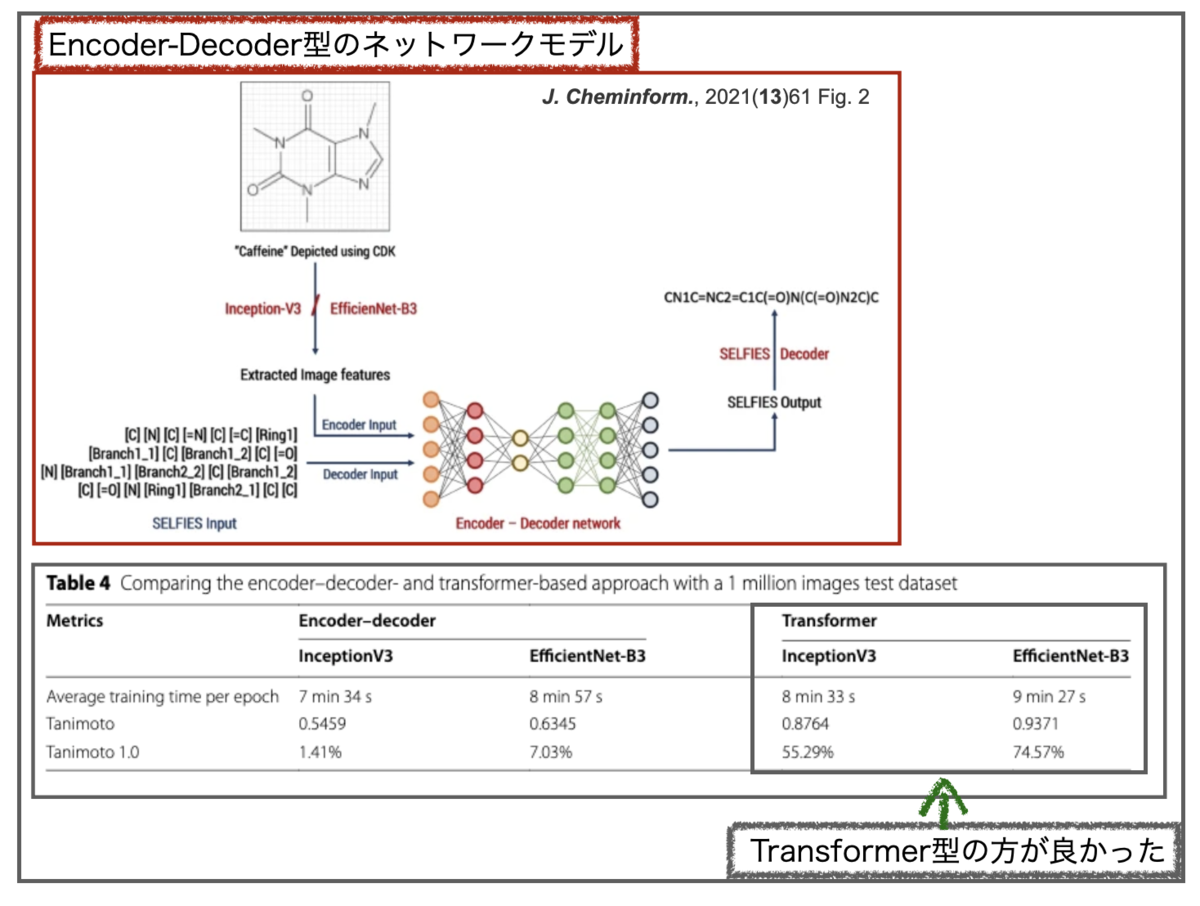
入力の画像認識 CNNのネットワーク構造としては、InceptionV3とEfficientNet-B3の2つを主に比較して、後者の方がパフォーマンスが良かった様です。
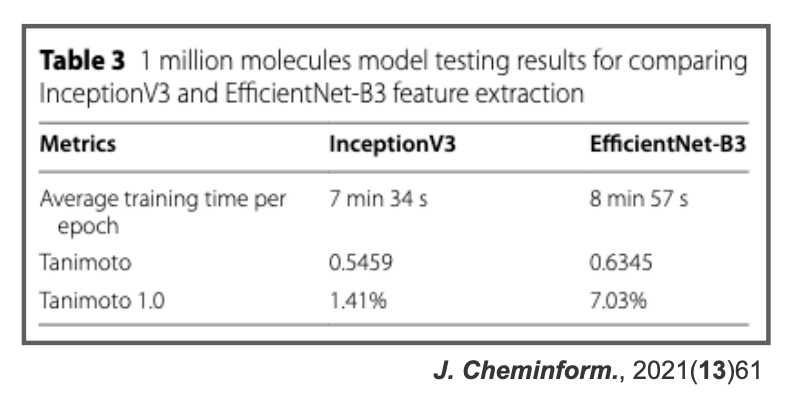
詳しくないのでよくわからないですが、各分野で最新の技術を色々と盛り込んだ感じでしょうか?最先端を攻める感じがアカデミックでいいですね!
1-3. DECIMER 1.0のデータセットと守備範囲
モデルの枠組みが大体わかったので、DECIMER 1.0の守備範囲についても参照しておきましょう!
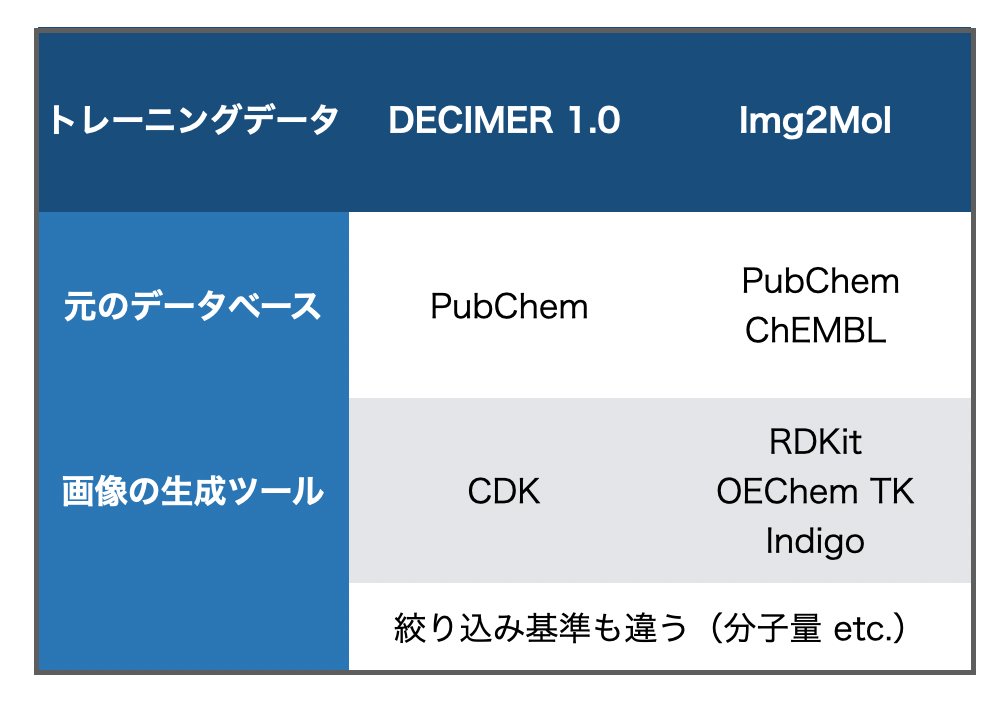
前回の記事では触れませんでしたが、Img2MolはPubChemとChEMBLから抽出し、一定の基準で絞り込んだ化合物をトレーニングに利用しています。また、トレーニング用の画像は複数のツール(RDKit、OpenEyeのOEChem TK、Indigo)を使って生成しています。
一方DECIMER 1.0は、対象データベースはPubChemのみ、トレーニング画像の生成はCDKのみを使用するといった、トレーニングデータセット準備段階での違いがあります。*2
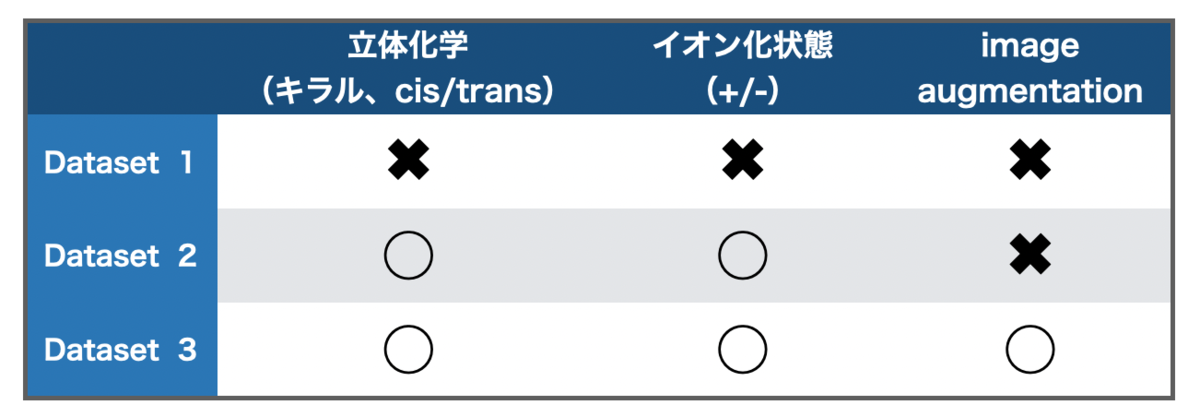
個人的に面白かった点は、DECIMER 1.0は「立体化学やイオン化状態を含めるとどうなるのかか?」という検証にも挑戦しているところです。
論文中で使われているデータセットは以下の様に「立体化学/イオン化の情報があるか?」、「Image augmentationをしたか?」で3つに分かれています。
以下のTable 8がDataset 1に対する結果、Table 12がDataset 2に対する結果です。データセットのサイズが近いsubset同士を比較すると、Table 12の方がパフォーマンス(Tanimoto、Tanimoto 1.0)が低下する傾向にあるようです。
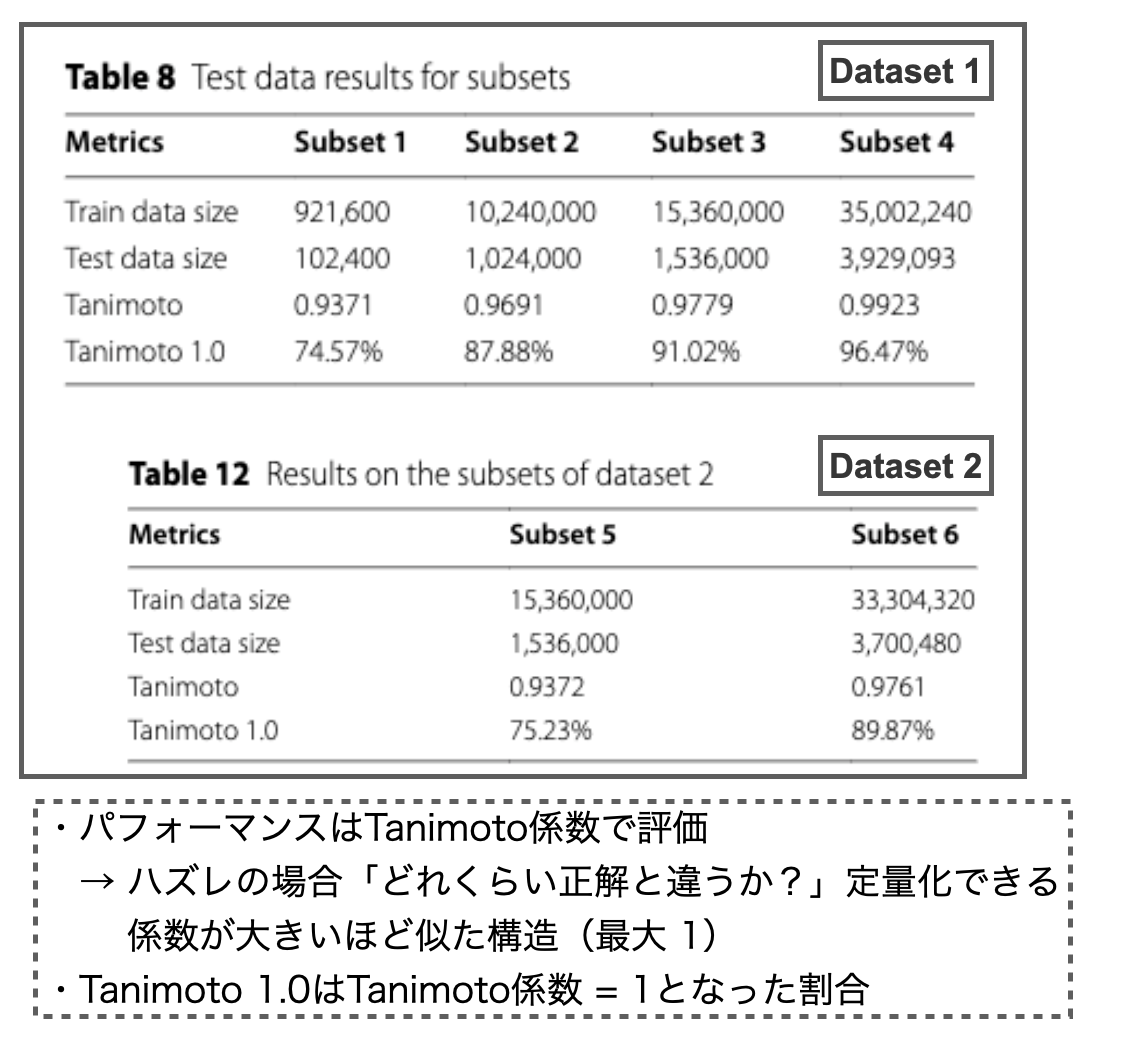
立体化学やイオン化状態の識別も考え始めると、問題が一気に複雑化するのでパフォーマンスが落ちるのは納得です。それでも結構良い結果が得られているように思いますが、皆さんはどう思われますか?
なおImage augmentationは、データ拡張を行ってDataset 2のデータを追加する(Dataset 3)とパフォーマンスが良くなるかを検証したものです。興味のある方は論文のFig. 10やTable 15あたりをご参照ください。
2. DECIMER 1.0で遊ぼう!
DECIMER 1.0の概要とImg2Molの違いがだいたい分かってきたので早速遊んでみましょう!
DECIMER 1.0はMITライセンスで公開されているので企業の方にも手を伸ばしやすいかもしれません。
後ほど記載しますが、学習済みパラメータはGoogle cloudのStorageにあり、プログラムを初めて利用するときにダウンロードする仕組みとなっているみたいです。
2-1. インストール
インストール方法はGitHubのREADMEの通りです。conda環境での利用が簡単なのでオススメとのことです。
# pipでgithubからインストール $ pip install git+https://github.com/Kohulan/DECIMER-Image_Transformer.git # もしくは以下でPyPiからインストールしてもOK $ pip install decimer
ところが、Img2Molの時と同様、Macユーザーはちょっとだけ面倒です。
MacOSでNvidia GPUがない場合は「tensorflow==2.3.0」を指定して入れなければいけないとの記載があります。上のコマンドでは私のMacにDECIMERをインストールできませんでした。
そこでgit cloneでDECIMERを落としてきた後、setup.cfgを少し書き換えるとインストールできました。
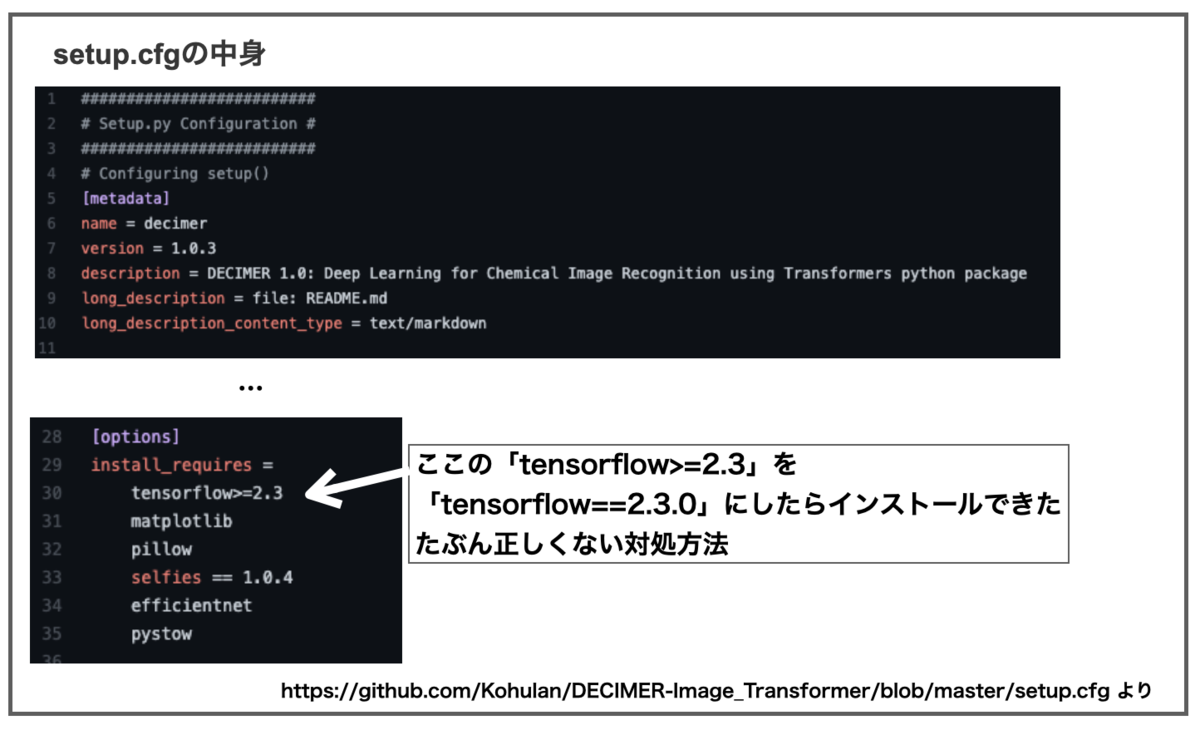
こういう感じの作業でした。
# githubからディレクトリを落としてくる $ git clone https://github.com/Kohulan/DECIMER-Image_Transformer.git decimer # setup.cfgを上図の様に書き換えて保存 # ディレクトリを移動 $ cd decimer/ # インストール開始 $ pip install -e.
どなたか正しい方法を教えてください。
2-2. 訓練済みパラメータの入手
先に書いた様に、DECIMER 1.0は学習済みパラメータも公開されていて、初回利用時にダウンロードされます。
私はパラメータの保存場所などが後でわからなくなるのを防ぐために、手動でダウンロードしてdecimerディレクトリの中に入れることにしました。
decimerのsrc/decimer/ディレクトリにあるDECIMER.pyをみると以下の記載があります。

デフォルトのディレクトリにパラメータファイルがなければmodel_urlからダウンロードしてくるみたいなことが書いてあります(たぶん)。
というわけでmolde_url = "https://~~~~" となっているURLにアクセスしてパラメータのzipファイルをダウンロードしました(1GBくらい)。
パラメータ保存場所として、DECIMERディレクトリにTrained_Modelsという新しいディレクトリをつくり、そこにzipを展開したファイルを格納しました。あとはDECIMER.pyやDECIMER_V1.0.pyの中にあるモデルのパスの箇所を書き換えて保存したら終わりです。
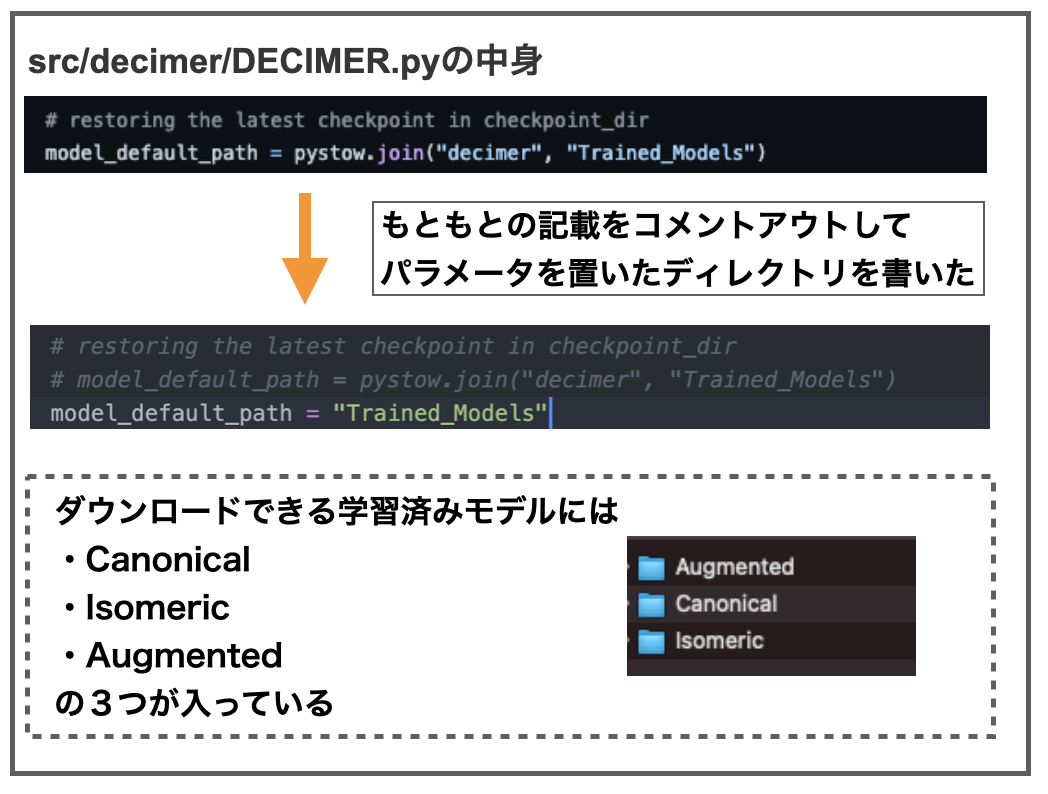
訓練済みのモデルにはCanonical、Isomeric、Augmentedの3つがあります。おそらく文献で触れられていた3つのデータセットに対応するものだと思います。*3
ダウンロードファイルの保存先を覚えておけばこんな余計なことをする必要はないと思います。私はすぐ忘れるんです。。。
何はともあれインストールとパラメータの準備ができました!
2-3. サンプルで遊んでみよう!
とりあえずJupyter notebookを起動して遊んでみましょう。Sample_Imagesディレクトリに画像例も準備してくださっているので利用します。
ライブラリをインポートしたあと、画像ファイルのパスと使いたいモデルを指定するだけでSMILESに変換して返してくれます。
from decimer import DECIMER # モデルの指定 model_name = "Isomeric" # 画像のパスを指定 img_path = "Sample_Images/caffeine.png" # 変換前に元々の画像を確認する from IPython.display import display from PIL import Image display(Image.open(img_path))
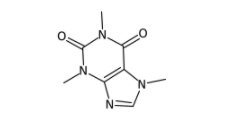
上のような画像を入力とします。さてDECIMERは無事SMILESに変換できるでしょうか??
# DECIMERでSMILESを予測 res = DECIMER.predict_SMILES(img_path, model_name) print(res) # > CN1C=NC2=C1C(=O)N(C)C(=O)N2C
SMILESができました!でもあってるのかわからない!読めない!
RDKitで変換した図をみてみます。(DECIMERと共存させられなかったので別のnotebookでやりました。)
from rdkit import Chem from rdkit.Chem import Draw m = Chem.MolFromSmiles("CN1C=NC2=C1C(=O)N(C)C(=O)N2C") Draw.MolToImage(m)
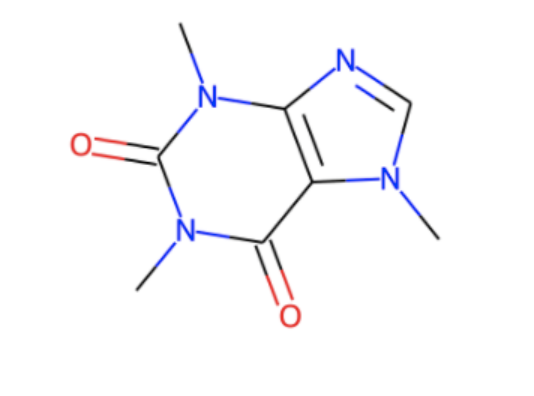
どうやらDECIMERはカフェインを認識してうまくSMILESに変換できたようです!
他のmodelではどうでしょうか?CanonicalとAugmentedで行った結果はこんな感じです。
AugmentedはIsomericと同様正しく認識できました!残念ながらCanonicalは全ての原子が硫黄の鎖状構造と誤認識してしまったようです。
論文を見る限りAugmentedが一番良さそうなので、以降の検証はこちらで行ってみます。
気になるのは立体化学の認識です。サンプルファイルに立体化学を含む例がありました。
どうだ!
おお!不斉点が認識されました!!SMILESに@が入っているだけでテンションが上がります!!
RDKitでの描画の都合上向きが変わってしまっていますが、立体化学も正しいようです。また、不斉の明記されていない炭素原子もありますが、ここも不斉情報がないものとして正しく認識できています。すごい!!
次に電荷を帯びたイオンはどうでしょうか??こんな例がサンプルにありました。
プラス電荷が認識されました!サンプルなのでうまくいくだろうとは思っていても実際に結果が出ると嬉しいですね!
話がそれますが、描画を並べてみるとRDKitの原子を色分けするスタイルはわかりやすくて良いですね。
2-4. 別のサンプルでテストしよう!
さてDECIMERの使い方がわかってきました。
次に気になるのはソフトの汎用性です。ソフトウェアのサンプル画像以外でもうまく機能するでしょうか???
比較対象としてちょうど良いのでImg2Molに提供されていたexamplesでテストしてみましょう!
カフェインに類似した構造の画像例です。
残念!だめでした。。。出力は芳香族がうまく認識できておらず、窒素-硫黄結合といった構造もできてしまっています。
別の例ではどうでしょうか?
こちらもだめでした。今回は芳香環を一つ認識できたましたが、なんとなく似ていそうで全然違う結果になってしまいました。
ひょっとして画像の化学構造式に色が含まれているのが悪かったりするのでしょうか?
別の画像例としてWikipediaのプロリンを使ってみましょう。元素記号が大きく、原子の色分けがない画像です。こちらもImg2Molではうまくいったものです。
なんだかすごい構造になりました。。。こんなアミノ酸は嫌だ。
最後に手書き画像の例を見てみましょう。Img2Molのサンプルです。
こんな結果になりました。
DECIMER 1.0の文献中にも記載がありましたが、環構造の認識に失敗すると、ヒトの目にはとても大きな間違いにみえますね。
上手くはいきませんでしたが手書きの構造式も化学構造として認識できいるという点はなかなかすごいと思います。
3. おわりに
以上、今回は化学構造式のOCRソフト DECIMER 1.0で遊んでみました。Img2Molと比較していかがだったでしょうか?
個人的には立体化学の認識、電荷の認識にも挑戦していて、サンプル画像ではうまく認識できているという点が驚きでした。Img2Molは立体化学を全て無視するようでしたのでDECIMERの長所だと思いました。
一方で、サンプル画像以外の画像では認識があまり上手くいっていないという印象でした。Img2Molは小さな分子であればWikipediaやPubChemのキャプチャ画像でも認識できたので、汎化性能(?)という点では課題があるかもしれません。
このあたりトレーニングデータセットの作り方による差もありそうです。Img2Molでは複数のツールで入力画像を作っていたのに対し、DECIMERはCDKのみでした。それぞれ異なる描画のクセがあるソフトを組みあせたデータセットの方がバラエティがあり、汎化性能が高くなりそうです。
DECIMER 1.0をImg2Molのデータセットでトレーニングしたらどうなるか?気になるところです。
あくまで素人の感想です。すみません。。。
また、DECIMERプロジェクトはImg2Molで今後の課題となっていたセグメンテーションにも既にWebアプリを公開しています。OCSRに関わる複数のソフト群全体の開発として取り組まれているので、シナジー効果によりさらに精度が改善していくかもしれません。次報が楽しみですね!
今回も色々と間違いが多そうです。ご指摘いただけると嬉しいです。
ではでは!
化学構造式のOCRソフト Img2Molで画像をSMILESに変換しよう!
少し前になりますがKaggleでBristol-Myers Squibb - Molecular Tranlationというコンペティションが開催されていました。
化学構造式を含む画像をその化学構造のInchi(International Chemical Idendifier)文字列に変換する精度を競うものでした。*1
巨大製薬企業のブリストル・マイヤーズスクイブがこのようなコンペティションを行うくらいなので、化学構造式のOCR(Optical Charactier Recognition)、「画像から化学構造式情報をコンピュータ処理できる形で抜き出すタスク」がニーズがありかつ難しいものであることが分かります。
さて今年のRDKit UGM 2021でも同様のタスクについての発表がありました。
その名もImg2Mol‼︎
- 発表の動画: YouTube : Djork-Arné Clevert: Img2Mol – SMILES Recognition from Depictions ofChemical Structure
- 発表のスライド:Clevert_Img2Mol.pdf
- 文献 : Img2Mol – accurate SMILES recognition from molecular graphical depictions Chem. Sci., 2021,12, 14174-14181
- GitHub : bayer-science-for-a-better-life/Img2Mol
そんな難しいタスクのためのプログラムが公開されている??
それなら遊ぶしかない!やってみましょう!
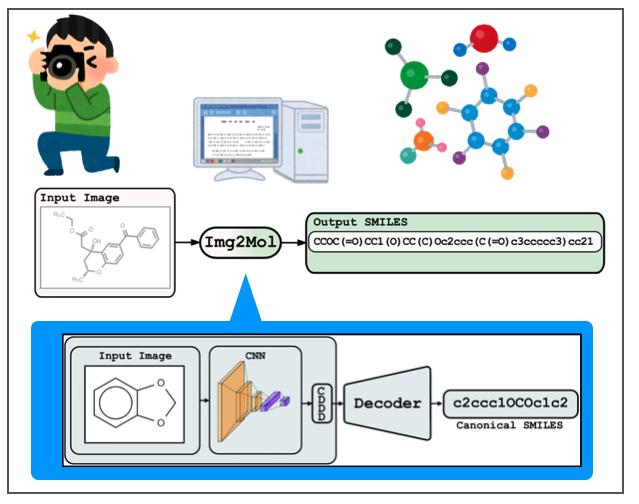
文献を流し読み
Img2Molはバイエルの機械学習チームが開発したプログラムで、化学構造式の画像をSMILESに変換してくれます。文献がオープンアクセスになっているのでざっと眺めてみます*2。
化学構造式OCRのモチベーション
そもそもなぜ化学構造式を画像から機械処理できる形で取り出すタスクに需要があるのでしょうか?
それは創薬化学の重要な情報源である文献と特許がともにコンピューター処理に適した形で情報を提供していないからです。
最近ではJournal of Medicinal ChemistryなどSupplementary Informationで化学構造のSMILESを列挙したファイルを提供するものも出てきましたが、まだまだ多くの文献で化学構造式は画像でのみ示されています。また特許では公開されている書類がPDFや画像形式であり、さらに産業上の理由から敢えて曖昧に書かれていることもあります。
このような状況なので、最新の情報を効率的に2次利用できる形でとりだしアップデートしていくのはとても大変です。というわけで画像から構造式をとりだすことにニーズがあるそうです。
ところで生理活性物質の公開データベースとしてみんな大好きなChEMBLも、専門家による手作業のキュレーションで情報更新されているようです。維持・運用のコストを考えると感謝しながら使わないといけませんね。
Img2Molのアプローチ
だいたいモチベーションがわかりました。では実際にタスクをこなすにはどうすれば良いでしょうか?
課題としてはこんな感じです。
1. 画像中の構造式を認識する
2. 認識した構造式をコンピューター処理できる形に取り出す
3. 取り出した構造は化学的に妥当なものである必要がある
1.と2.は全てのOCRに共通で、3.は化学ならではの課題です。
で、この化学ならではの部分をどうするか?
Img2Molでは画像を畳み込みニューラルネットワーク(CNN)で認識し、化学構造式の特徴を表現する空間(CDDD)に一度埋め込んだあと、線形表記(Canonical SMILES)として取り出す、という2段階の構成をとっているようです。

CDDDはcontinuous and data-driven molecular descriptorsの略で、同じくバイエルのチームが開発した記述子です。こちらの論文もOpen Accessになっています。
- Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations Chem. Sci., 2019,10, 1692-1701
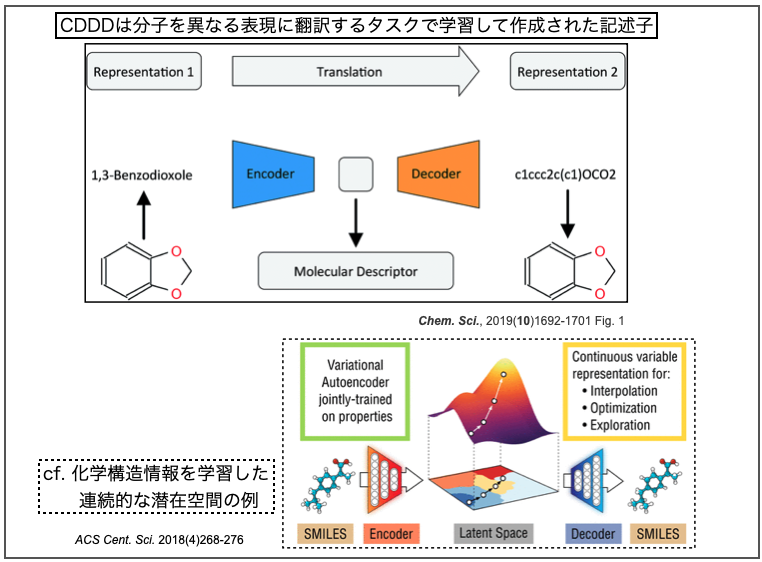
オートエンコーダ(Autoencoder、自己符号化器)はニューラルネットワークの一つで、「入力されたデータを一度圧縮し、重要な特徴量だけを残した後、再度もとの次元に復元処理をするアルゴリズム」だそうです*3。
ある化学構造を表すSMILESには複数通りの書き方があり、それらを入力として(一意の)canonical SMILESを出力する翻訳のタスクをこなすオートエンコーダを学習します。このネットワークは化学構造の特徴を学習した潜在空間(latent space)となっています。なので、化学構造を表す記述子として使うことができ、異なるタスクにも色々と応用できるよ!(とかそんな話です。たぶん。)
上図で引用した下半分はAspuru-Guzik先生のチームによる以下の論文です。*4
- Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules ACS Cent. Sci. 2018, 4, 2, 268-276
ところで、バイエルのチームはImg2Molと同様のアプローチを以前にも報告しています。以下の論文では「ECFP記述子を入力として、もともとの分子構造を逆に推定するリバースエンジニアリング」を行っています。
- Neuraldecipher – reverse-engineering extended-connectivity fingerprints (ECFPs) to their molecular structures Chem. Sci., 2020,11, 10378-10389
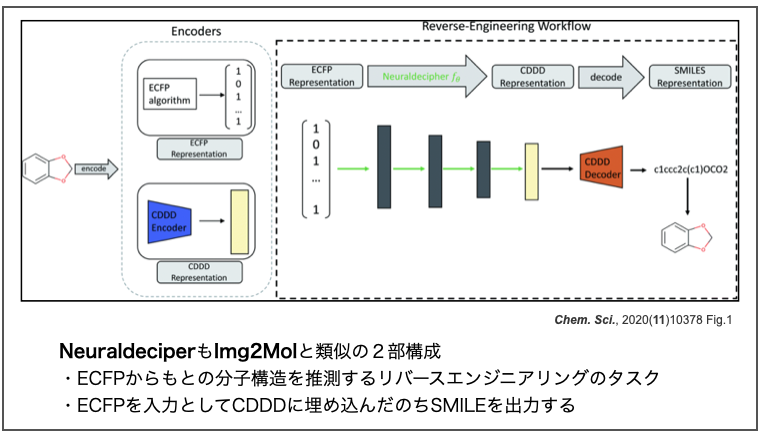
このタスクでも一度CDDDに埋め込んだ後SMILESとして出力しています。応用範囲が広い記述子を開発できると便利だなー。
Img2Molのパフォーマンス
Img2Molのネットワーク構造やメソッドの詳細はよくわからないので割愛します。ごめんなさい。
結果っぽいのだけ貼っておきます。
他の手法よりパフォーマンスが良かったよの図(ベンチマーク)。
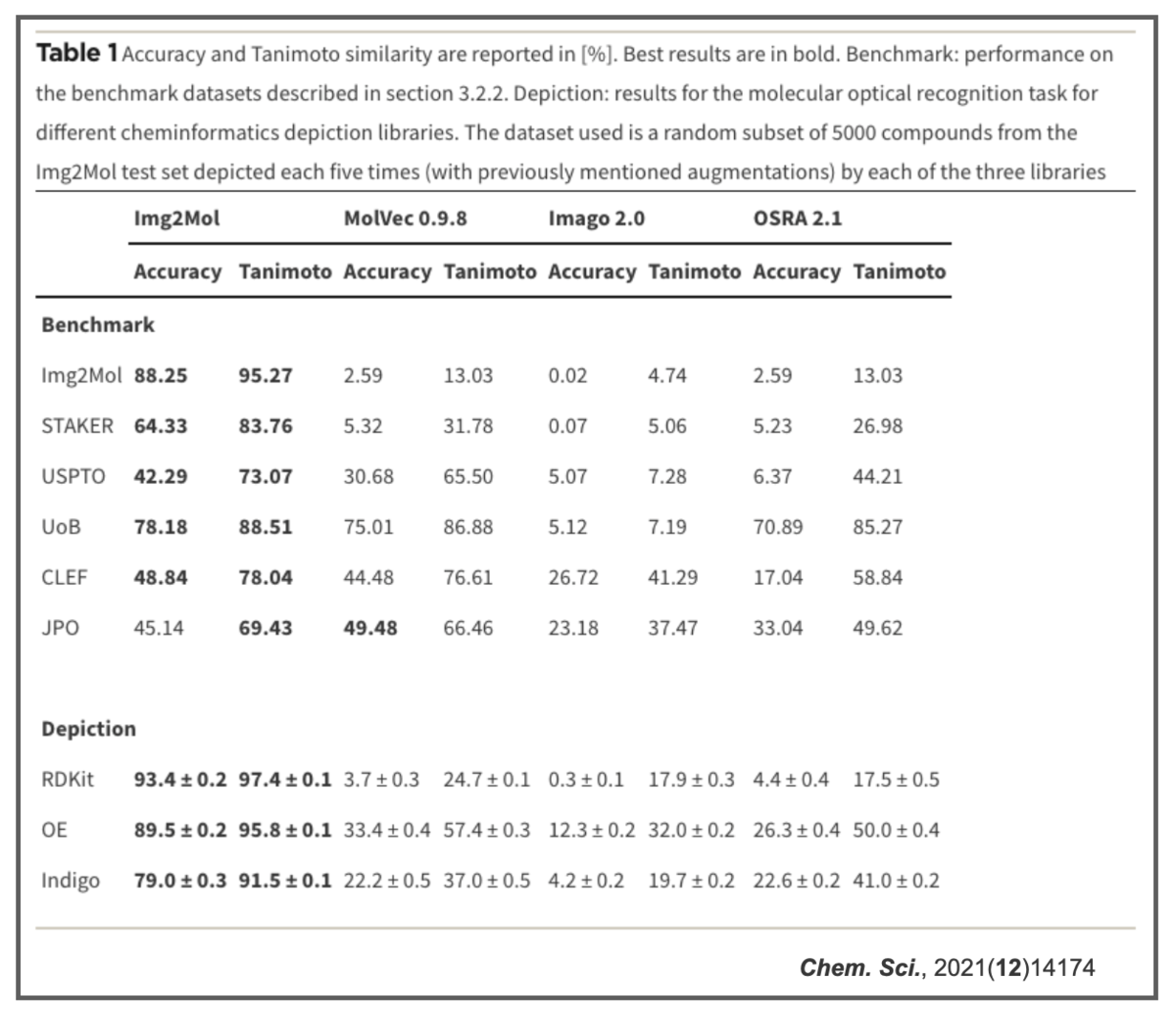
AccuracyだけでなくTanimoto類似度でも評価しているのが面白いです。
学習に使う化学構造を描くツール(depiction)によっても結果が変わっているのも興味深いです。同じ化学構造でもソフトによって表現・相性が変わるところに、コンピュータで化学を扱う難しさを感じます。
さらに面白い結果はこちら。画像の解像度(Resolution)とパフォーマンスの関係です。なんとImg2Mol (no aug)は入力の解像度が上がると精度が落ちています。
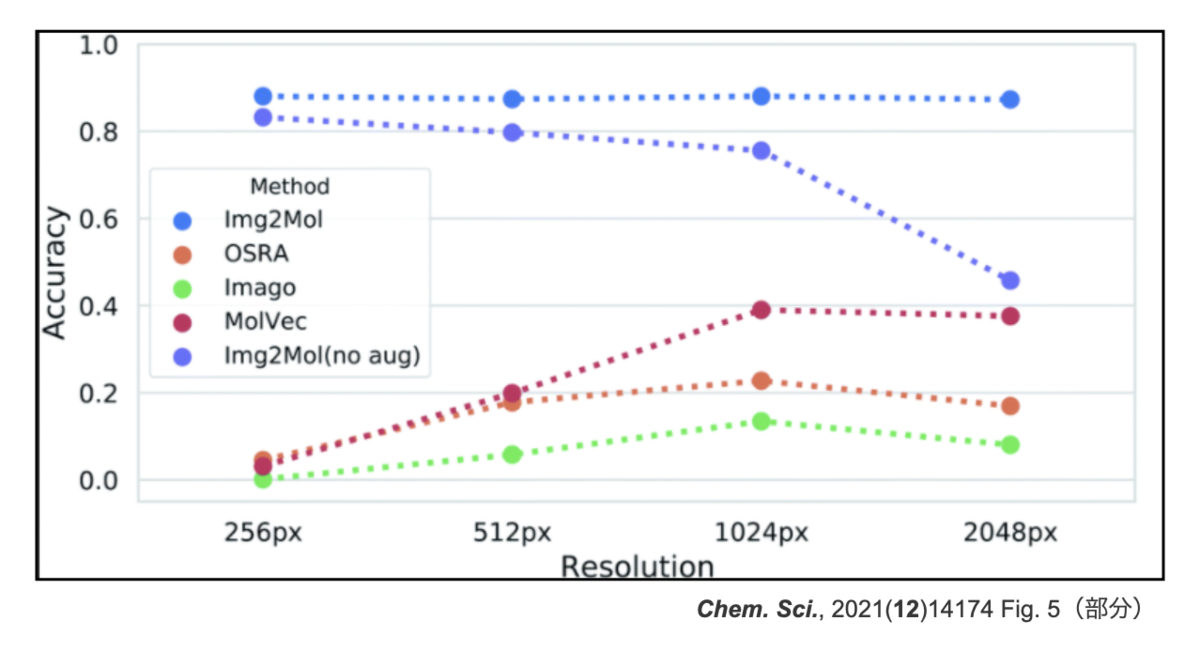
「入力の質が上がって精度が悪くなるの??逆じゃないの??」となりますが、どうやらネットワークへの入力の仕方によるもののようです。
ネットワークの構造は決まっているので、入力画像は全て同じ形(224 x 224 px)にリサイズされます。高解像度の画像は、このリサイズの処理で失われる情報が逆に多くなるため、ぼんやりとした入力になってしまうようです。
この解像度の問題や分子サイズの大きな構造の認識が難しいという問題を解決するため、data augmentationをおこなって学習し直しています。これで得られたよりロバストなネットワークが上図の「Img2Mol(無印)」に相当するものです。高解像度でもパフォーマンスが落ちていません。
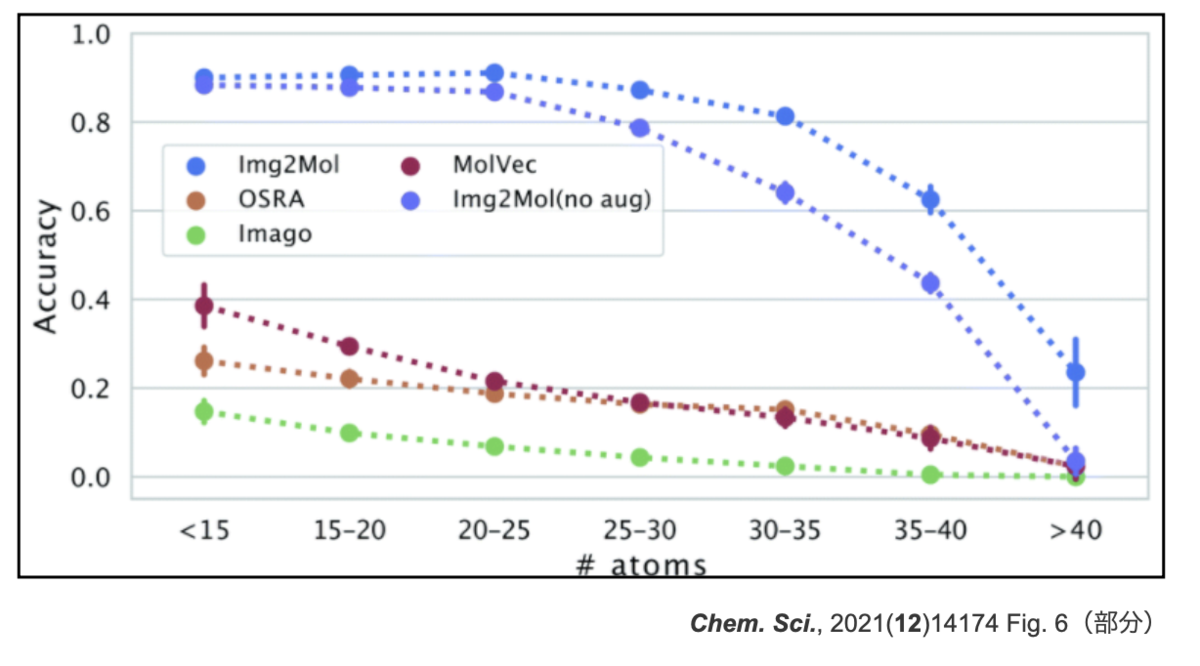
分子サイズとパフォーマンスとの関係も貼っておきます。こちらもaugmentationで改善はしていますが、原子数が30を超えたあたりから精度が落ちてきています。
以上、Img2Molのざっとした文献紹介でした。正確な情報は論文やRDKit UGMの発表動画などをご参照ください。
Img2Molで遊ぼう
それでは早速、Img2Molで遊んでみましょう!素晴らしいことにGitHubで公開してくださっています!
但し、学習済みパラメータのライセンスはCC BY-NC 4.0でnon-commercial use onlyですので商用利用できないことには十分ご注意ください(念のため)。*5
インストール
インストール方法はcondaでOKで簡単です。GitHubのREADMEの以下をコピペするだけ。
git clone git@github.com:bayer-science-for-a-better-life/Img2Mol.git cd Img2Mol conda env create -f environment.yml conda activate img2mol pip install .
・・・が、Macユーザーの方はご注意ください。Img2Molに必要なソフトとして「cudatoolkit=11.0」の指定がありますが、NVIDAがmacOSのサポートを終了したらしく、「CUDA ToolKit v11」はmac版がありません。*6
Img2Molのインストールに使う「environment.yml」ファイルは以下のようになっています。

このままだと「condaチャンネルにそんなの無いよ!」って言われます。なので「cudatoolkit=11.0」のバージョン指定「=11.0」を消して「cudatoolkit」だけにして上書き保存してから再実行するとインストールできました。
旧バージョンのCUDA ToolKitが入るので動作の保証はできませんが、とりあえず動いたのでヨシ!*7
学習済みパラメータファイルはGoogleDriveからダウンロードできるようにしてくださっています(→ https://drive.google.com/file/d/1pk21r4Zzb9ZJkszJwP9SObTlfTaRMMtF/view )。大きい(~2.4GB)ので空き容量とインターネット回線にご留意を!
「model.ckpt」というファイルがダウンロードされるので、Img2Molディレクトリの中の「model」ディレクトリの中に放り込めば準備完了です。
Exampleで遊ぼう
準備ができたらJupyter notebookを開いて遊びましょう!
使い方がexample_inference.ipynbに、画像例がexamplesディレクトリにあるのでそのまま使わせていただきます。
# 必要なライブラリのインポート import torch from img2mol.inference import * from IPython.display import display # img2molを使う準備 device = "cuda:0" if torch.cuda.is_available() else "cpu" img2mol = Img2MolInference(model_ckpt="model/model.ckpt", # パラメーターファイルの指定 device=device) cddd_server = CDDDRequest() # > Initializing Img2Mol Model with random weights. # > Loading checkpoint: model/model.ckpt # > Setting to `self.eval()`-mode. # > Sending model to `cpu` device. # > Succesfully created Img2Mol Inference class.
これでImg2Molを使って画像からSMILESを予測するためのクラスが作成できました!
あとは変換したい画像のファイルパスを指定すれば予測を実行できます。
# exaplesディレクトリのdigital_example1.pngファイルを入力 res = img2mol(filepath="examples/digital_example1.png", cddd_server=cddd_server)
結果は辞書型で以下のようになっています。
print(type(res)) # > <class 'dict'> res # > {'filepath': 'examples/digital_example1.png', # > 'cddd': None, # > 'smiles': 'Cn1c(=O)c2c(nc(Sc3ccccc3)n2C)n(C)c1=O', # > 'mol': <rdkit.Chem.rdchem.Mol at 0x7fb09587eda0>}
入力ファイルとCDDD、結果のSMILESとRDKit Molオブジェクトも入っています。
今回入力に使ったのは以下の画像。
input_img = Image.open(res["filepath"], "r") display(input_img)
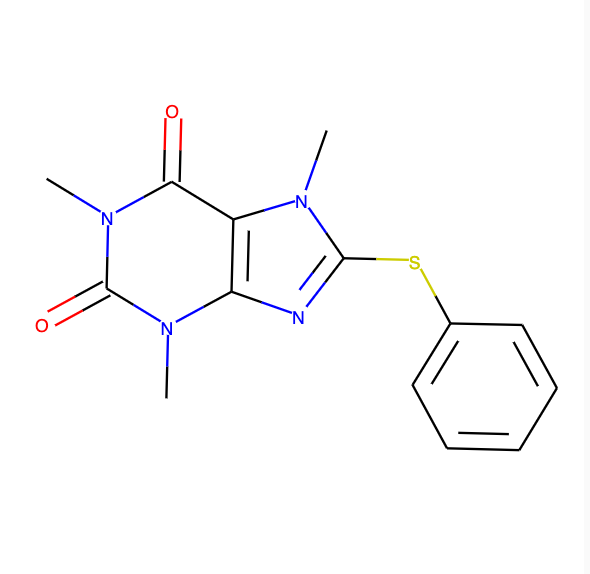
予測結果はこんな感じ。RDKit Molオブジェクトなのでjupyter notebookで構造式をそのまま描画できます。
# 変換後のSMILESを表示 print(res["smiles"]) # > Cn1c(=O)c2c(nc(Sc3ccccc3)n2C)n(C)c1=O # RDKit Molオブジェクトの構造式を描画 res["mol"]
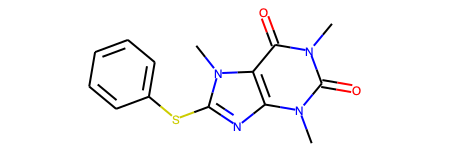
おお!正しく認識できてます!
私はSMILESを読めないのでわかりませんが構造式で確認できるので便利!!
手書きの構造式の例も用意されています。こういうのです。
# handwritten_example2.pngファイルを入力 res = img2mol(filepath="examples/handwritten_example2.jpg", cddd_server=cddd_server) input_img = Image.open(res["filepath"], "r") # 入力画像の確認 display(input_img)
結果はこんな感じ
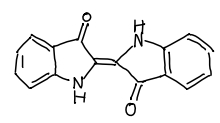
# 変換後のSMILESを表示 print(res["smiles"]) # > O=C1C(=C2Nc3ccccc3C2=O)Nc2ccccc21 # RDKit Molオブジェクトの構造式を描画 res["mol"]
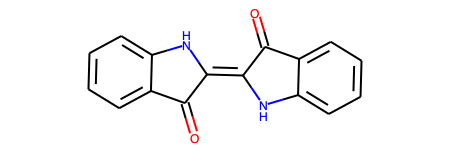
こっちもうまく認識できています!手書きの認識ができるなら授業のメモの電子化にも使えそうです!すごい!!!
ちなみに失敗例も提供されてたりします。コードは同じなので省略します。
こんなのです。
真ん中の環構造がフェニルではなくインドールとして認識されています。入力画像の右側ヘテロ環の結合関係がうまく認識できていないようです。
手書きの構造式は結合の長さや角度がまちまちだったり、元素記号のアルファベットが崩れていたりと、色々難しいポイントが多そうです。
いろいろな構造で遊ぼう
使い方が分かったので色々な画像を認識させてみましょう!
今年も色々な分子が話題となりましたが、なんといってもノーベル化学賞が有機触媒の開発に贈られたことは一大ニュースでしたよね!
というわけでプロリン画像をWikipediaから持ってきました(Wikipedia-プロリン)。こんな結果です。
うまくできました!
マクミラン触媒はどうでしょうか?PubChemの構造を使ってみます((5S)-5-Benzyl-2,2,3-trimethylimidazolidin-4-one)。こんな感じです。
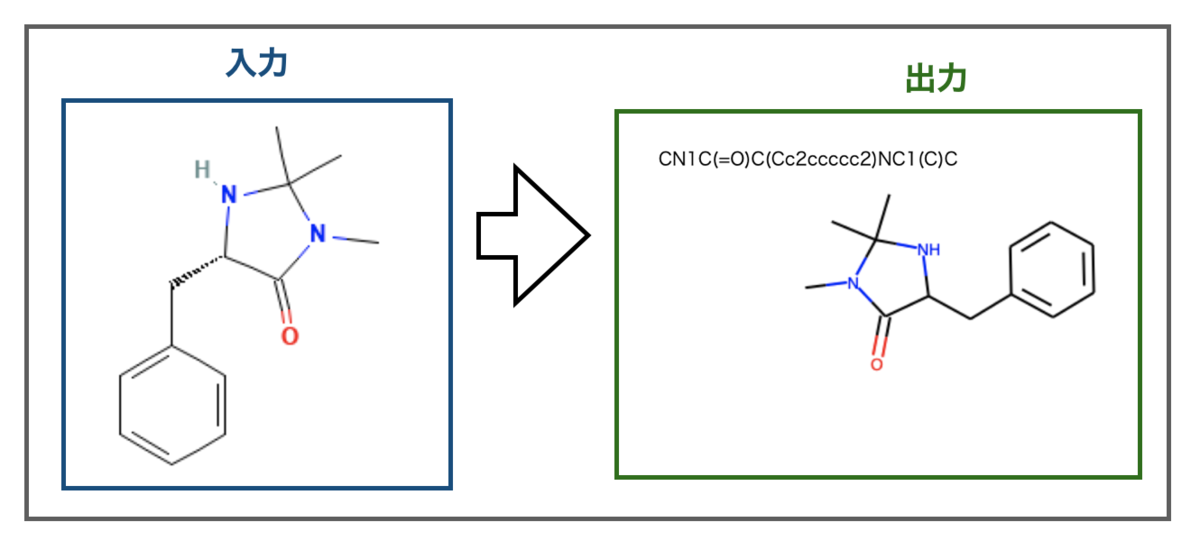
上手くいきました・・・が、不斉の情報までは認識できていないようです。出力のSMILESにキラル情報(@文字)がなく、構造式も不斉がありません。
ぱっとみた感じCDDDの学習で不斉の取り扱いまでは行っていないようなので、不斉情報は記述子に埋め込まれていないのかもしれません。
ところで化学構造の描画方法には今まで見てきたもの以外にも色々あります。Ball and StickやSpace-Fillingといったモデルで化学構造を表した画像はどのように認識されるのでしょうか?
同じくPubChemから画像を作成してImg2Molに投げてみました。
Ball and Stick描画したマクミラン触媒はこんな結果になりました。
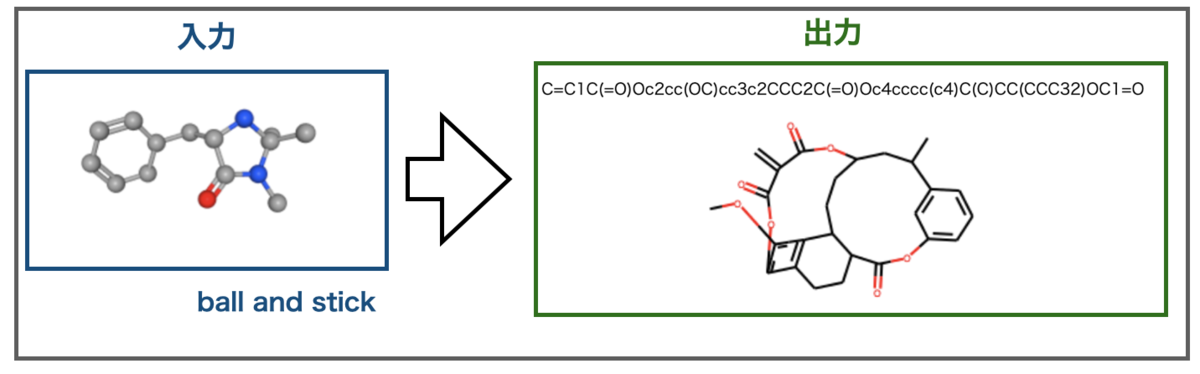
すごい構造。。。出力には窒素原子が含まれていませんが、色で原子を区別した描画では区別できないのでしょうか?
次にSpace-Fillingで描画したマクミラン触媒はこんな結果でした。
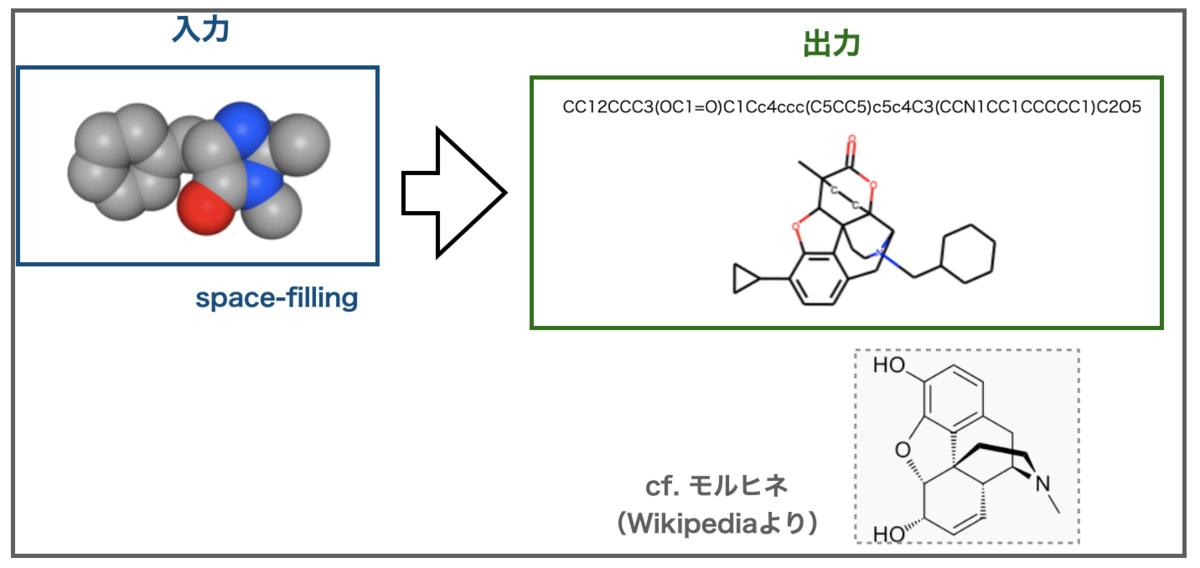
面白いことにSpace-Fillingの出力はモルヒネ様の骨格に認識されました。さらに今度は窒素原子も入っています???
出力に含まれる酸素原子の数を考えると、色で窒素を認識できていると思えないのでモルヒネ様に誤認識して引きづられた結果かもしれません。Img2Mol内部でどの様に認識されているのでしょうか???
不適切な入力構造はここまでにして。。。。
さて論文中では分子のサイズ(原子数)が大きくなると精度が落ちるとの記載がありました。最後に試してみましょう!
今年話題になった分子といえばKRAS G12C阻害剤として承認されたAmgenのSotorasibがありますね(Wikipedia - Sotorasib)。組成式C30H30F2N6O3なので、水素原子を除いても原子数41となります。
さてどうなるでしょうか?
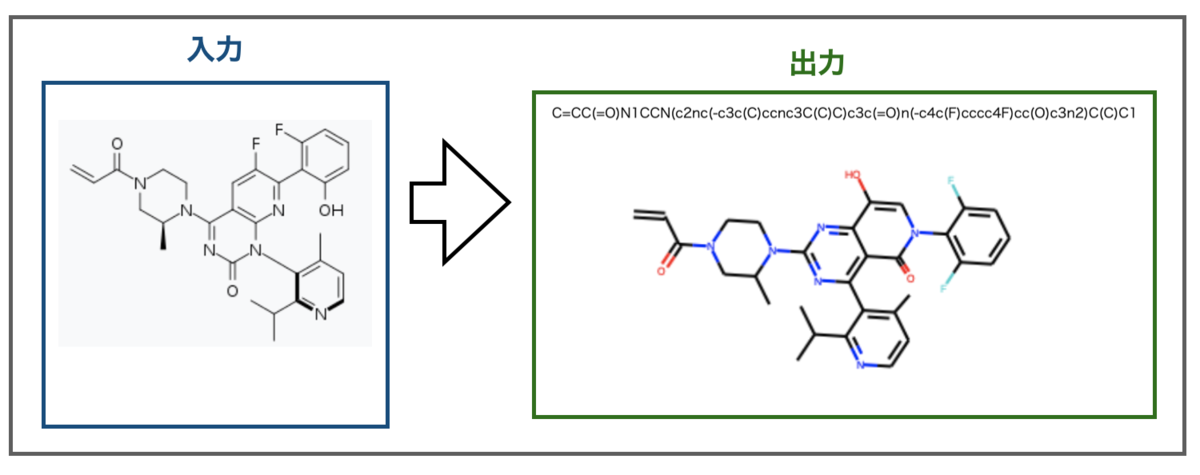
ぱっと見認識できている様にも見えますが、ちょっとずつ違います。
骨格の真ん中の環構造はカルボニルの位置や他の環との接続の仕方が異なります。また右端のフェニル基はオルト位に置換があるのは良いもののどちらもフッ素原子になってしまっています。入力画像の真ん中の環のフッ素原子と位置が近いので誤認識したのかもしれません。
画像からの構造の読み取りが失敗したとしても大枠が正しいなら細かい修正だけで済むので良いかな?とも思いましたが、微妙にちょっとずつ違うと間違い探しみたいで逆に難しくなってしまうかもしれません。
課題
いくつか遊んでみた結果、かなり精度が高く便利なツールという印象ですがいくつかできないこともわかってきました。
- 不斉の認識
- 分子サイズの大きなもの
また、利用を想定されていないであろう描画方法(ex. Ball and Stick, Space-Filling) 、色で表された原子の識別はできませんでした。
化学構造の特徴を学習した潜在空間にうつすならひょっとしていけるかも…と思いましたがダメでした。無茶を言ってすみません。
この他にRDKit UGMの動画で指摘されていたものにはキャプションつきの画像がありました。化学構造式以外の情報(化合物名など)が同じ画像の中にあると区別して認識できないとのこと。
「画像のセグメンテーションをどうするか?」が次の課題のようです。
おわりに
以上、今回は化学構造式のOCRソフト、Img2Molで遊んでみました。課題はあるもののかなり便利なツールだなぁと思いました。
ところで国語の成績も悪かった私ですがいまだに覚えている短歌があります。
「のみ込めぬままに図表は消されゆき 遠き席にて聴き終えて立つ」
詠み手*8が学生の時、講義の内容が理解できないうちに板書が消されてしまい、そのまま講義が終了してしまって「あーあ、やんなっちゃったよ」みたいな内容だった気がします。
なぜ覚えているか???
同じ経験をたくさんしたからです!
そう!化学構造式が超高速で現れては消えていく有機化学の授業でね!!
Img2Molがあればパシャーと写真撮って、後で画像からいい感じで構造式を抽出できたかもしれません。。。
今回も色々と間違いが多そうです。特に論文の中身。ご指摘いただけると嬉しいです。
おしまい。
*1:DeNAのデータサイエンティストの方々が参加されたチームが3位入賞・金メダルを獲得されたそうです(DeNA News)。格好良い!
*2:CC BY-NC 3.0
*3:オートエンコーダ(Autoencoder)とは|意味、仕組み、種類、活用事例を解説
*4:CDDD文献中にも引用されています
*5:ちなみのコードのライセンスはApache-2.0です。但しcddd_server.pyもCC BY-NC 4.0なので注意が必要です。
*6:NVIDIA、macOSのサポートを正式に終了した「CUDA Toolkit v11」をリリース。MacではCUDAアプリの開発と実行は出来ない状態に
*7:ネットワークの学習も自分でやろうとしたら不具合が出るのかもしれません。いつも通り無理矢理押し切っているのでおすすめしません。