医薬品と回転異性体 〜実例とちょっと計算の検証〜
こちらは創薬アドベントカレンダーに間に合わなかった記事です。毎度、初歩の初歩の記事で恐縮ですが折角なのでここに供養します。
さて、クイズ!以下の4つの医薬品!構造も作用機序も全然違いますが、ある共通点があります!一体なんでしょうか??
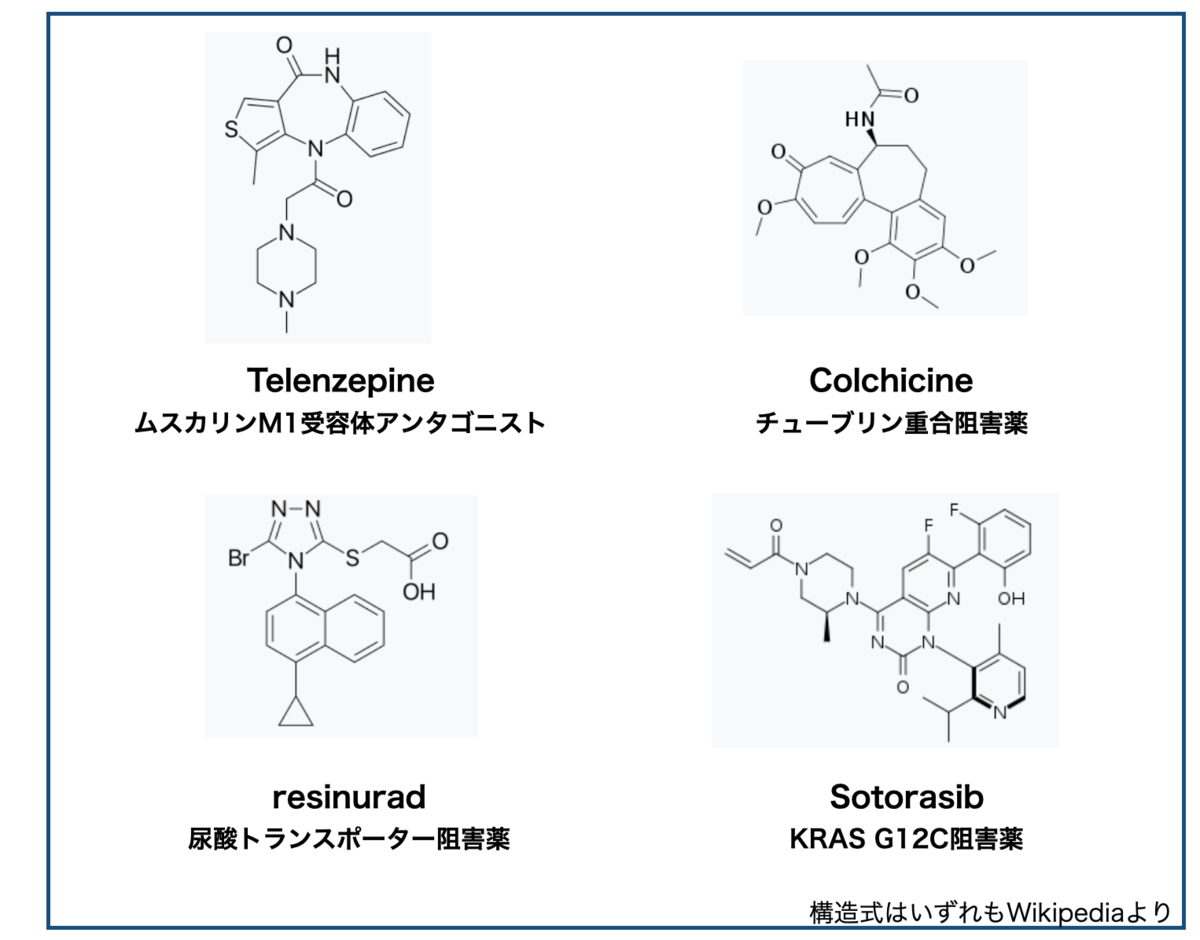
答え・・・「回転異性体(アトロプアイソマー)をもつ医薬品」。タイトルでネタバレしてましたけどね!
正直、私は構造式を見ただけではどこに回転異性が潜んでいるか分かりません。ということで、回転異性体判別力(?)をあげるべくエネルギー変化をシミュレーションして遊んでみたいと思います。
1. 回転異性と医薬品
まずは基本的な背景知識のおさらいです。
1-1. 回転異性の復習
低分子医薬品において不斉(キラリティー)は重要な要素です。2021年ノーベル化学賞「不斉有機触媒」の解説でも、不斉合成の重要性の例として医薬品が引き合いに出されていました。
この不斉の一つ、軸不斉は「分子が不斉中心を持たないが、キラリティー軸を有するキラリティーの特別な形式の一つ」です(wikipedia)。
軸不斉の例として回転異性(アトロプ異性)があり、回転異性を持つ化合物の例としては野依先生が合成方法を確立されたBINAPがあります*1。
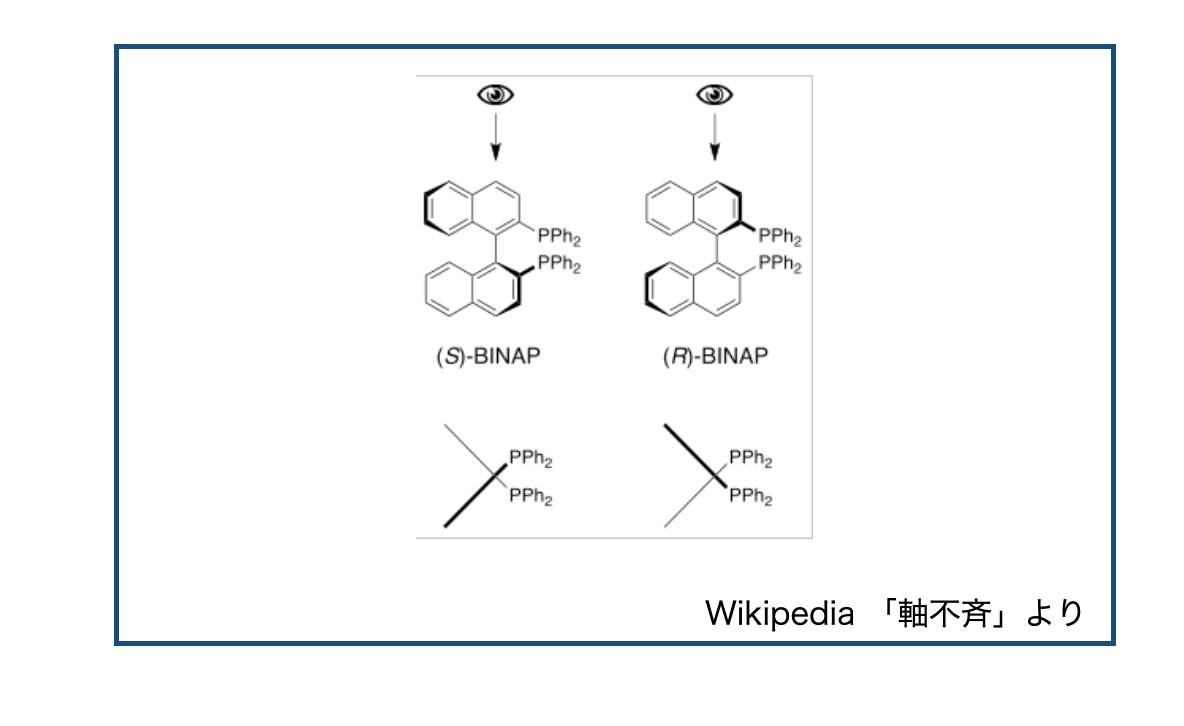
要するに、立体障害が大きくて自由回転できないから鏡像異性体ができちゃうぜってやつです。*2
1-2. 冒頭の例ではどこ?
「4つ異なる置換基がついた炭素」みたいな分かりやすい不斉中心がないので、回転異性体の有無はパッと見では分かりにくい気がします。
冒頭に挙げたそれぞれの化合物の不斉に関する軸はここです。
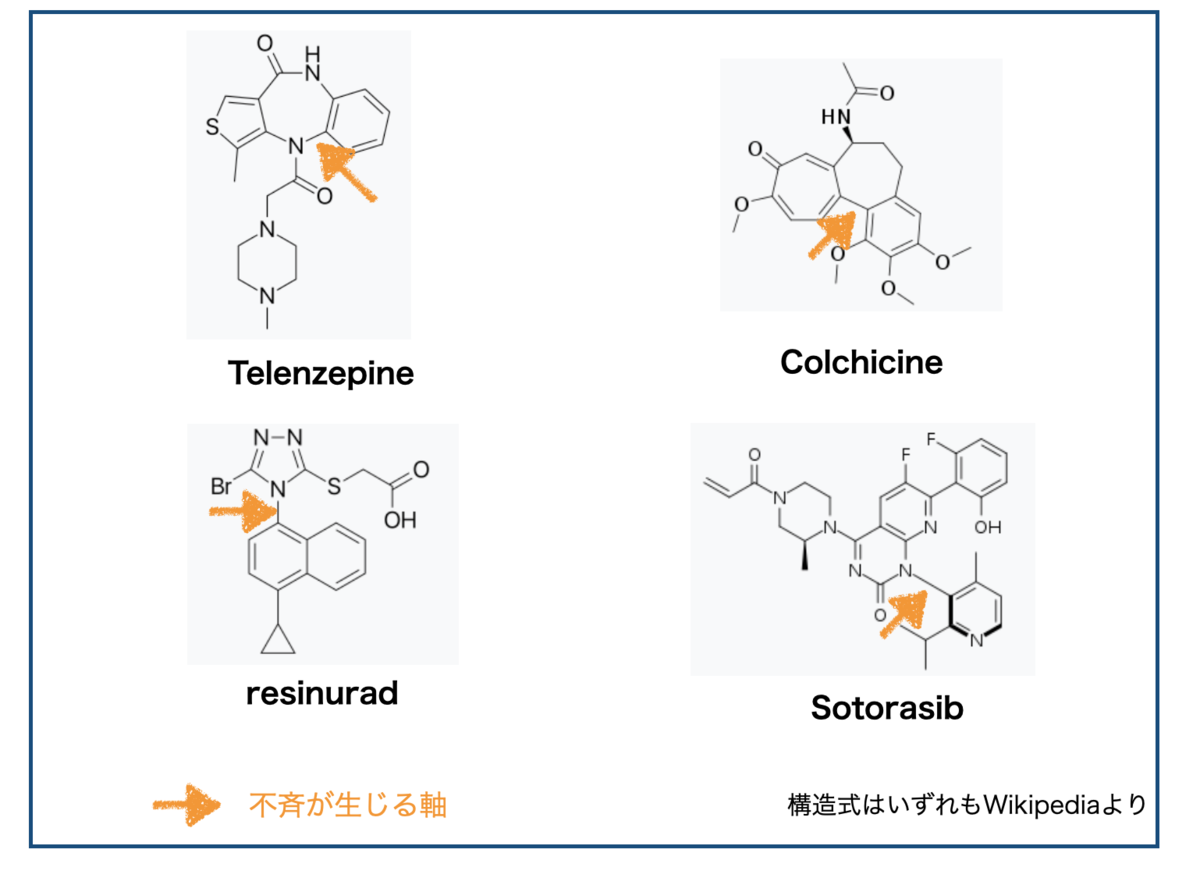
いずれも(ヘテロ)アリールに結合する箇所です。BINAPと見比べると雰囲気がわかってきました。
ちなみにWikipediaによるとTelenzepineは(+)-isomerと(-)-isomerでムスカリン受容体に対する活性に500倍以上差があるそうです(ラット大脳皮質)。鏡像異性体ってやっぱり生理活性に重要なんですね。
1-3. 回転異性体の安定性と分類
「回転異性体で活性差が大きいなら、良い方のだけ医薬品にすれば良いんじゃない?」となりますが、そんなに簡単ではないそうです。なぜなら、回転異性体の中には安定に分離できるものとできないものがあるからです。
回転異性体は軸周りの回転障壁の高さに由来します。障壁が十分に高ければ、異性体間での変換が十分に遅い(実質、変換しないとみなせる)ので、分離してそれぞれ別の化合物として扱う事ができます。一方で、障壁が中くらいであれば異性体を分離したところで、しばらくするとまた混ざってしまうので分離する意味がありません。
厳しい条件をクリアして医薬品として開発・臨床応用するためには、その化合物の均一性の保証も大事な要素です。生理活性も異なる2つの異性体が作るたびにバラバラに混ざってしまったら困ってしまいます。
以下の様に、回転障壁の高さに応じて回転異性体の安定性はざっと3つに分類できるそうです。(障壁のエネルギーは目安)*3*4
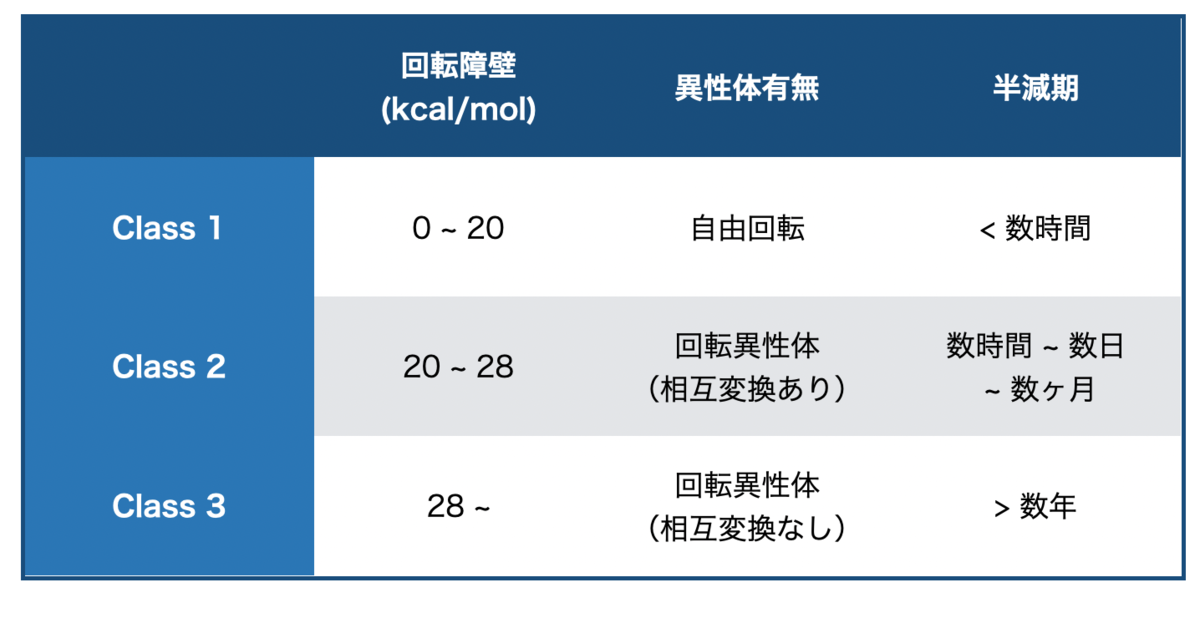
エネルギーの低いClass 1であれば、そもそも区別できるほど安定な回転異性体が生じないので一つの化合物として取りあつかえます。また、障壁が高いClass 3であれば年単位で安定に取り扱えます。面倒なのが中間のClass 2で、場合により、単体あるいは混合物として取り扱うようです。
1-4. 回転異性体への対処例
実際のところ、研究過程で回転異性体がでてきたらどうするのでしょうか?今年FDAに承認されたAmgen社のKras G12C阻害薬 Sotorasibの発見に至る経緯が面白いので参照しておきます。
- Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors
J. Med. Chem. 2020, 63, 1, 52–65
オープンで読めます!*5
研究の途中で見つかった化合物 18は有望な性質を持っていたものの、回転異性体がみつかりました。障壁は26 kcal/molで、25℃での半減期が8日。先の分類でいうとClass 2にあたりそうです。*6

回転異性体に対する戦略として著者らがあげているのは以下の3つです。
- ① もっと回りにくくして安定な異性体を分離できる様にする(Class 3にする)
- ② 逆に回りやすくして回転異性体が生じない様にする (Class 1にする)
- ③ 分子を対称形にして、回転しなくても鏡像異性体にならないようにする
検討の結果、戦略①がうまくいったそうです。軸不斉近傍にメチル基を導入することで回転障壁が30 kcal/molより大きくとなった化合物 24を見出しています。ここからさらに発展させて最終的にSotorasibを見出しています。
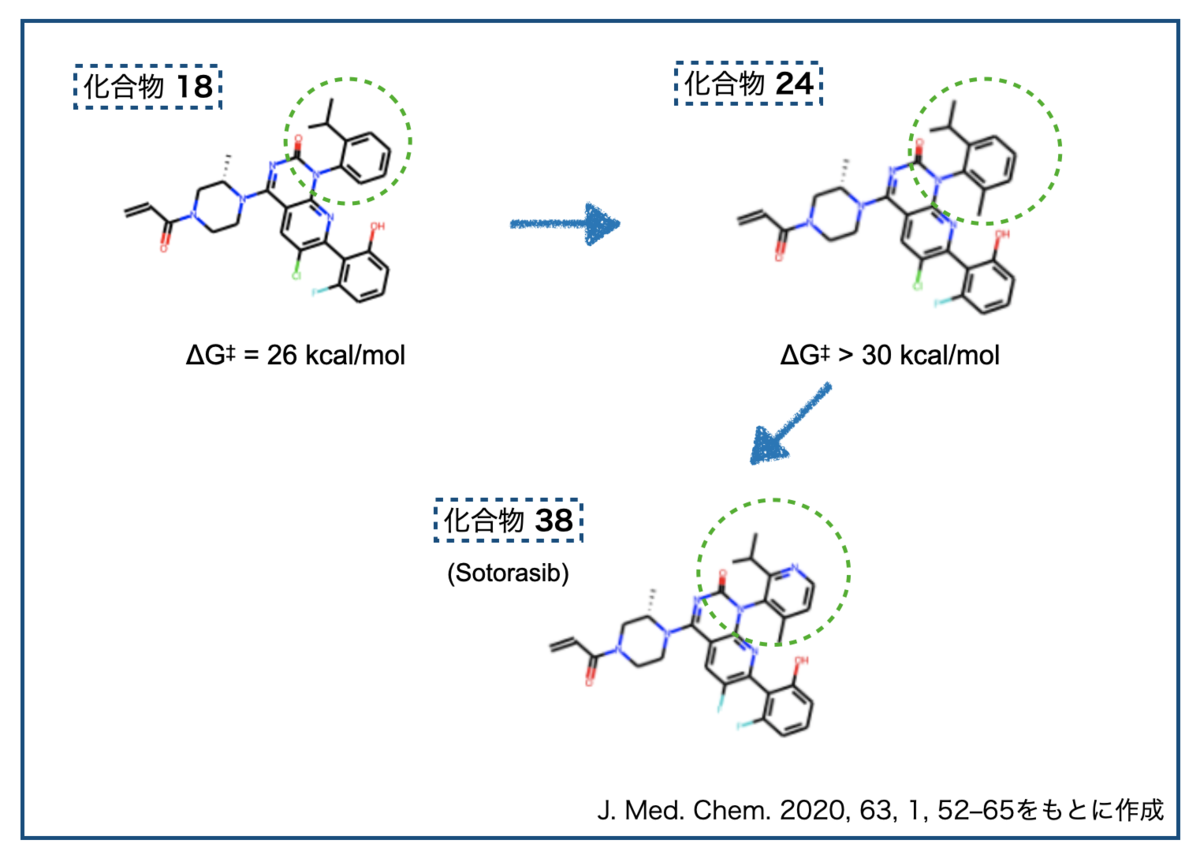
メチル基一つで回転異性という現象を制御してしまうの格好いいですね!承認された医薬品の構造式だけを見ても分かりませんが、そこに至る経緯を辿ると科学者の戦いが垣間見えて楽しいです!
2. 2面角とエネルギー変化
前置きが長くなりました。
構造式だけを眺めるだけではイマイチよくわからない回転異性体ですが、軸周りに回転させたエネルギー変化をたどれば感触がつかめるかもしれません。オープンソースのツールを使ってシミュレーションしてみましょう!
試す手法は以下の2つです。
- ① RDKitのコンフォマー生成による解析(Molecular Mechanics)
- ② xTBの2面角スキャンによる解析(Semiempirical Extended Tight-Binding)
以下の2つの記事で解説してくださっている内容を参考にさせていただいています。というよりほぼコピペです。ごめんなさい。
- 参考記事1:化学の新しいカタチ:RDKitを用いて制約付きで立体構造を生成する
- 参考記事2:Spinning coral : Torsion angle scan with rdkit & xtb
まず試すのは以下のモデル化合物です。先のJ .Med. Chem. の文献を参考にしています。
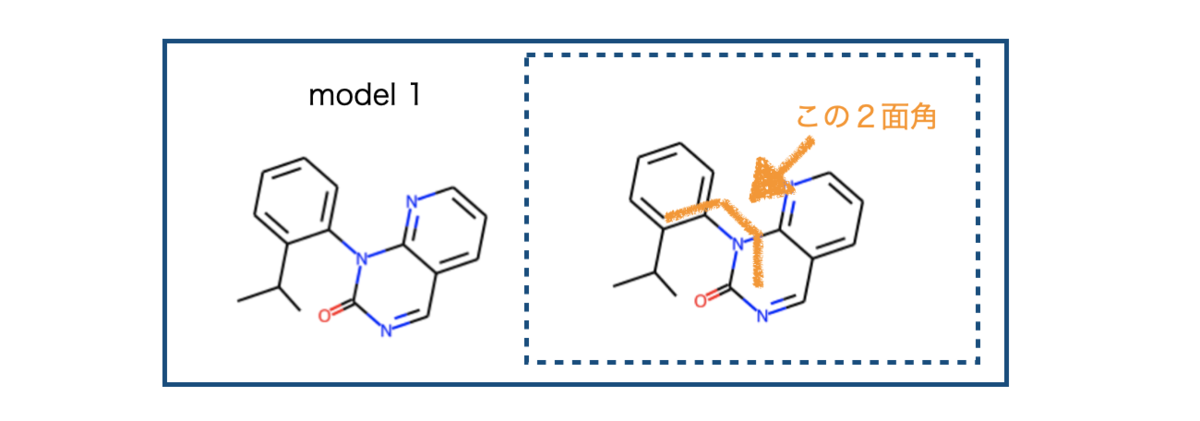
それでは2面角に依存してエネルギーがどのように変わるか? 眺めてみましょう!
2-1. モデル化合物 1の描画とatom indexの確認
まずはモデル化合物 1を描画して必要な情報を確認します。Molオブジェクトを作成し、考慮する二面角のatom indexをチェックします。
from rdkit import Chem from rdkit.Chem import AllChem, Draw # モデル構造作成 model_1 = Chem.MolFromSmiles("CC(C)c1ccccc1n3c(=O)ncc2cccnc23") # 水素原子付加 model_1H = AllChem.AddHs(model_1) Draw.MolToImage(model_1H)
RDKitのブログ記事より、drawMolDraw2DでaddAtomIndices=Trueとすることでatom indexを確認できます。*7
from rdkit.Chem.Draw import rdMolDraw2D from IPython.display import SVG d2d = rdMolDraw2D.MolDraw2DSVG(350,300) d2d.drawOptions().addAtomIndices=True d2d.DrawMolecule(model_1H) d2d.FinishDrawing() SVG(d2d.GetDrawingText())
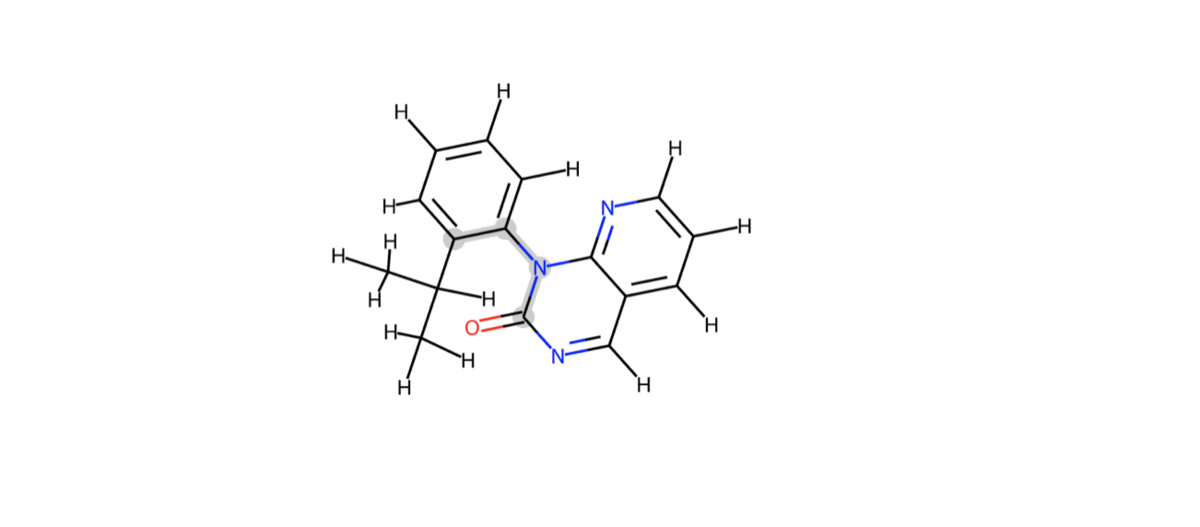
イソプロピル基の根元から軸周りのatom indexを今回の標的とします。小さくて見づらいですが3, 8, 9, 10が目的の2面角を定義する原子のatom indexです。
ハイライトして描画してみます(highlightAtoms)。
cp = Chem.Mol(model_1H) d2d = rdMolDraw2D.MolDraw2DSVG(350,300) d2d.drawOptions().setHighlightColour((0.8,0.8,0.8)) d2d.DrawMolecule(cp,highlightAtoms=[3, 8, 9, 10]) d2d.FinishDrawing() SVG(d2d.GetDrawingText())
結合(bond index)についても同じことはできます(addBondIndices=True)。見やすさのため水素原子を付加せずに確認します。
d2d = rdMolDraw2D.MolDraw2DSVG(350,300) d2d.drawOptions().addBondIndices=True d2d.DrawMolecule(model_1) d2d.FinishDrawing() SVG(d2d.GetDrawingText())
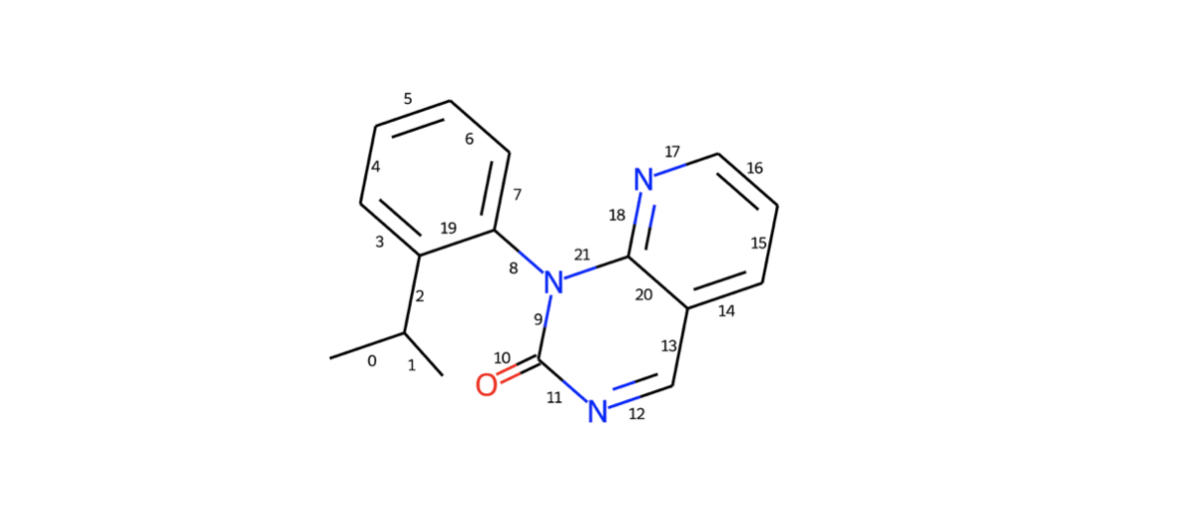
着目部位のbond indexは19, 8, 9でした。
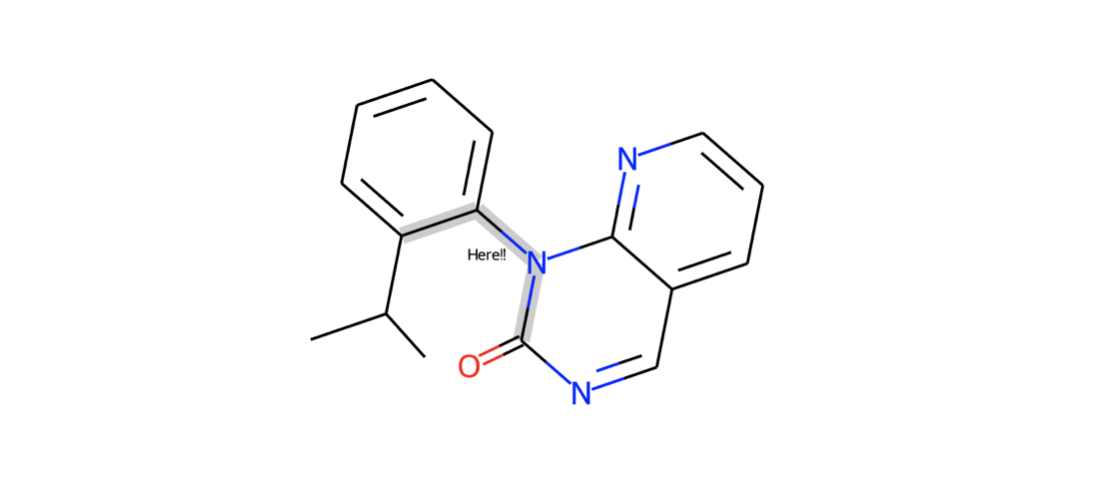
結合にコメントをつけて描画する事もできます(bondNote)。こんな感じ。
cp = Chem.Mol(model_1) cp.GetBondWithIdx(8).SetProp("bondNote","Here!!") d2d = rdMolDraw2D.MolDraw2DSVG(350,300) d2d.drawOptions().setHighlightColour((0.8,0.8,0.8)) d2d.DrawMolecule(cp,highlightAtoms=[],highlightBonds=[19, 8, 9]) d2d.FinishDrawing() SVG(d2d.GetDrawingText())
atom indexはSetAtomMapNumを使っておくと途中でズレたりせず安心だそうです*8。
また、これを使うと追加の描画の設定をしなくてもatom indexが表示されます。
for i, a in enumerate(model_1H.GetAtoms()): a.SetAtomMapNum(i) Draw.MolToImage(model_1H)
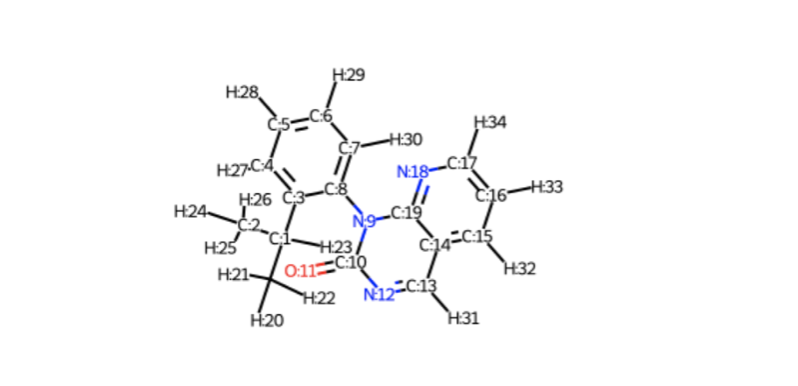
脱線しましたがatom indexがわかりました。
2-2. RDKitの制約付きコンフォマー生成
モデル化合物のMolオブジェクトとatom indexがわかったので、コンフォマーを生成してエネルギー変化を確認します。
目的の2面角に、180°を10°ずつ回転した制約を加えたコンフォマーを作成します。各コンフォマーはMMFF力場で構造最適化します(分子力学法)。
# コード参考元 1; https://future-chem.com/rdkit-constrained-conformer/ # コード参考元 2; https://pschmidtke.github.io/blog/torsion/dihedral/oss/opensource/rdkit/xtb/energy/2021/02/16/torsion-angle-scans-xtb.html # ベースになる3次元構造を生成(再現性のためrandomSeedを設定) AllChem.EmbedMolecule(model_1H,randomSeed=10) # ベースのコンフォマーを取得 conformer=model_1H.GetConformer(0) # ベースが更新されない様にdeepcopyしておく import copy m_1 = copy.deepcopy(model_1H) # MMFF力場の設定 m_1_p = AllChem.MMFFGetMoleculeProperties(m_1) # エネルギーを格納するリスト energy=[] # 回転する角度の範囲 angles=range(0,180,10) # コンフォマーのID = 0からスタート confid=0 # 角度範囲でforループを回す for angle in angles: # MMFF力場 ff = AllChem.MMFFGetMoleculeForceField(m_1, m_1_p) # 目的の2面角に制約を設定(atom indiex : 3, 8, 9, 10) ff.MMFFAddTorsionConstraint(3,8,9,10, False, angle - .1, angle + .1, 10000.0) # 構造の最適化(minimize) ff.Minimize() # エネルギーを取得してリストに追加 energy.append(ff.CalcEnergy()) # 次の新しいコンフォマーを準備 ## 新しいコンフォマーID confid+=1 ## 原子数を引数で指定して新コンフォマーを作成 new_conf = Chem.Conformer(model_1H.GetNumAtoms()) ## 新コンフォマーに原子の位置を設定 for i in range(model_1H.GetNumAtoms()): new_conf.SetAtomPosition(i, (m_1.GetConformer(-1).GetAtomPosition(i))) ## 新コンフォマーにコンフォマーIDを設定 new_conf.SetId(confid) ## 新コンフォマーをベースに追加 model_1H.AddConformer(new_conf)
print(model_1H.GetNumConformers()) # 19
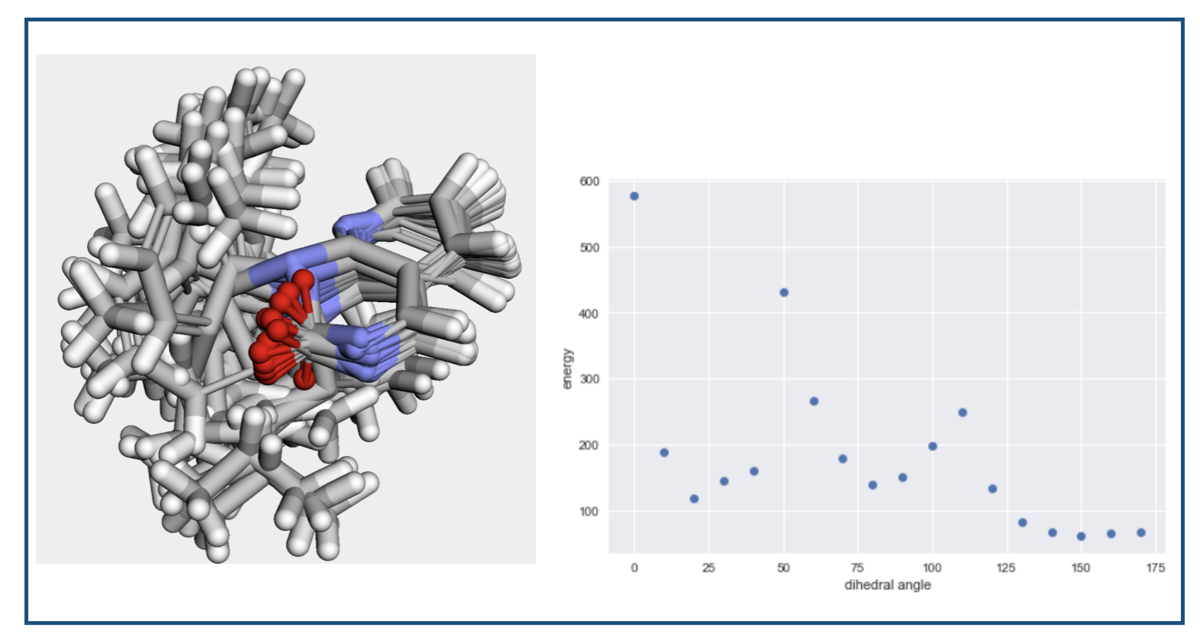
ベースとなるコンフォマーと、0° 〜 170° の10° 刻み18個の合わせて19個が含まれています。アザキナゾリノン骨格をコア構造にしたアラインメントを取って3次元で眺めてみましょう。
# 3次元を眺めるためにpy3Dmolを使う from rdkit.Chem.Draw import IPythonConsole import py3Dmol # 重ねたいコア構造をatomIdで指定してコンフォマーのアラインメントをとる AllChem.AlignMolConformers(model_1H, atomIds=[9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19]) # 3Dビューワーに表示(ベース以外の18個) v = py3Dmol.view(width=600, height=600) for cid in range(1, 19): IPythonConsole.addMolToView(model_1H, confId=cid, view=v) v.setBackgroundColor('0xeeeeee') v.zoomTo() v.show()
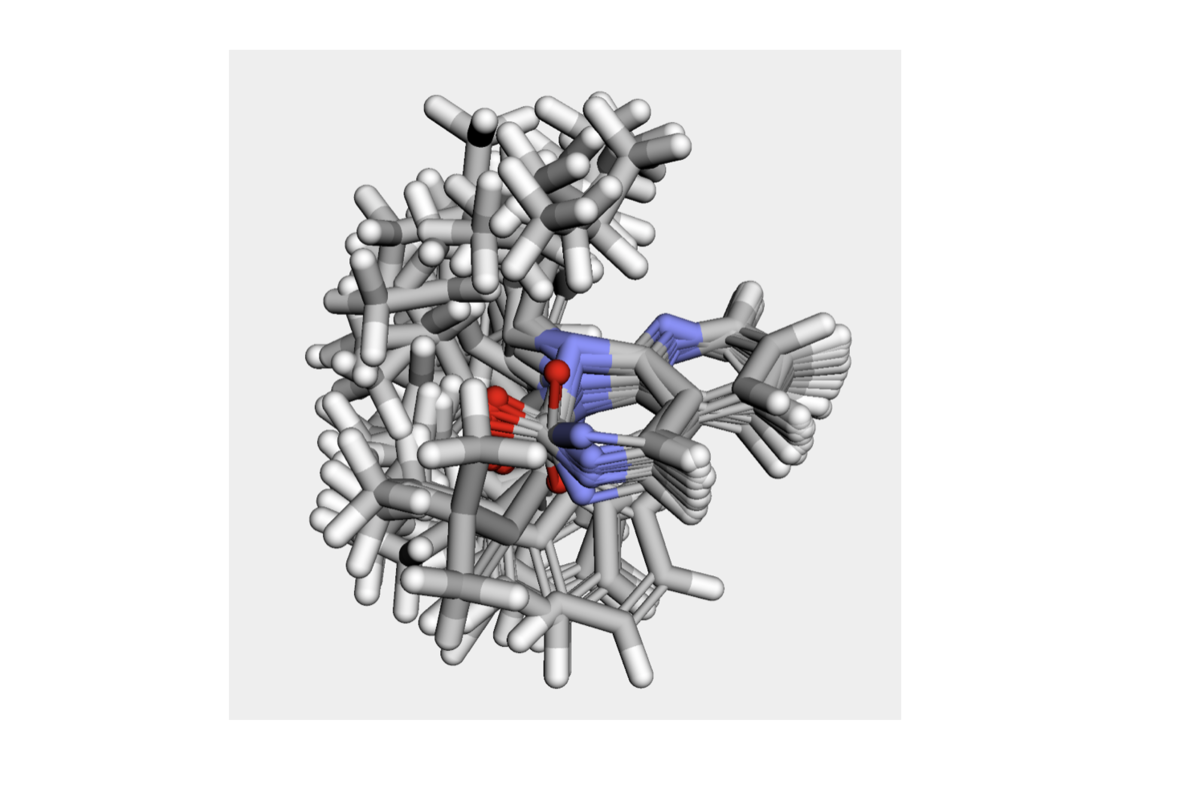
ぐちゃぐちゃですが、とりあえず見たい2面角周りの回転はできていそうです。アザキナゾリノン骨格の平面性がところどころ崩れていることに不安はありますが。。。。
角度とエネルギーの関係をプロットしてみます。
# matplotlibの設定 import matplotlib as mpl import matplotlib.pyplot as plt mpl.style.use('seaborn') # 角度とエネルギーの関係 plt.plot(angles, energy, 'o') plt.xlabel('dihedral angle') plt.ylabel('energy')
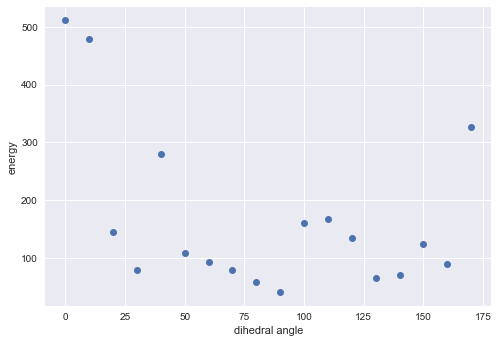
なるほど。よくわからん。。。
2-3. RDKitの制約付きコンフォマー生成~part 2~
先程3Dを眺めた際に骨格の平面性が崩れている感じがありました。どうもこれはRDKitのバージョンによって挙動が違うようです。
参考記事2によるとMMFFのout of plane項の設定に問題があるそうです。記事のおすすめに従ってSetMMFFOopTerm(False)を使ってみましょう。
オプションの設定以外は同じコードです。
# molオブジェクト作り直し model_1 = Chem.MolFromSmiles("CC(C)c1ccccc1n3c(=O)ncc2cccnc23") model_1H = AllChem.AddHs(model_1) AllChem.EmbedMolecule(model_1H,randomSeed=10) conformer=model_1H.GetConformer(0) m_1 = copy.deepcopy(model_1H) # MMFF力場の設定 # さっきとここが違う!Out of plane項の設定を変える。 m_1_p = AllChem.MMFFGetMoleculeProperties(m_1) m_1_p.SetMMFFOopTerm(False) energy=[] angles=range(0,180,10) confid=0 for angle in angles: ff = AllChem.MMFFGetMoleculeForceField(m_1, m_1_p) ff.MMFFAddTorsionConstraint(3,8,9,10, False, angle - .1, angle + .1, 10000.0) ff.Minimize() energy.append(ff.CalcEnergy()) confid+=1 new_conf = Chem.Conformer(model_1H.GetNumAtoms()) for i in range(model_1H.GetNumAtoms()): new_conf.SetAtomPosition(i, (m_1.GetConformer(-1).GetAtomPosition(i))) new_conf.SetId(confid) model_1H.AddConformer(new_conf)
新しく作ったコンフォマーを重ね合わせて眺めてみます。(コードは省略)
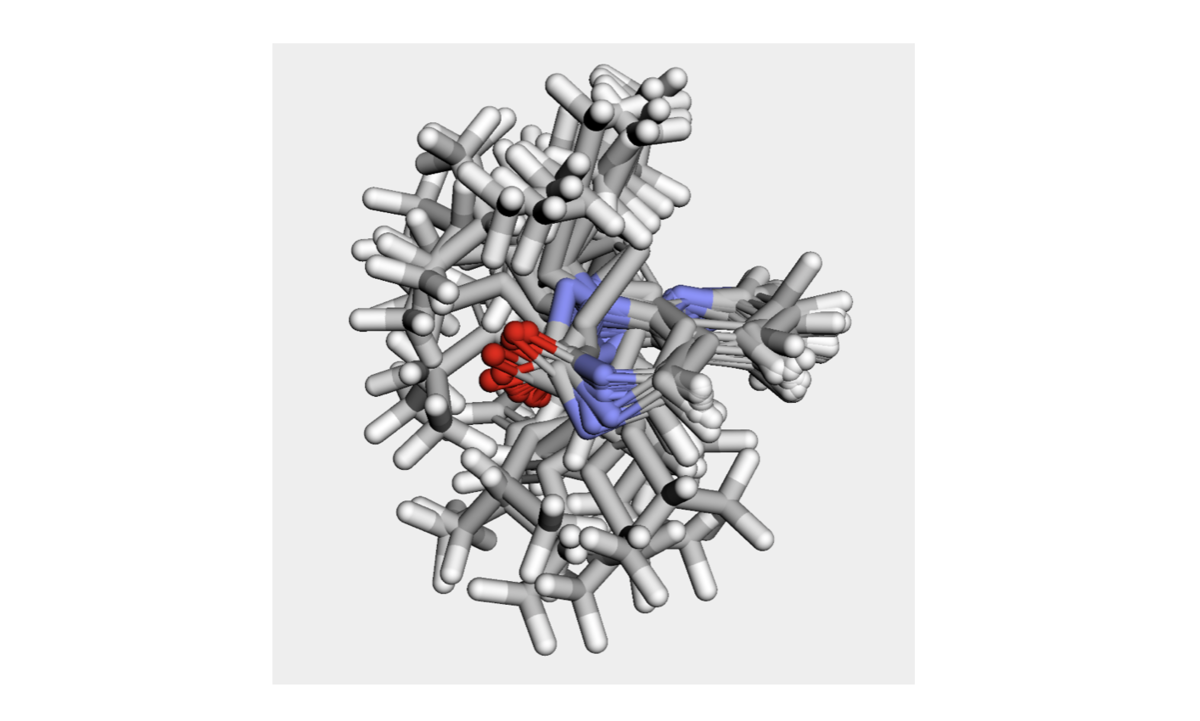
悪化しました。平面性が全然保たれていなさそうです。
一応、こっちでも角度とエネルギーの関係をプロットしてみました。
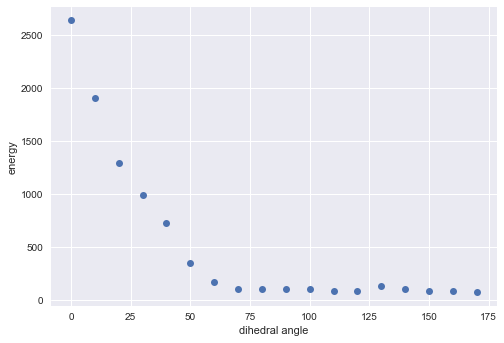
構造が崩れているので参考にはなりませんが、SetMMFFOopTerm()の設定で結果が大きく変わることは分かりました。
2-4. xTBによる2面角スキャン
RDKitのコンフォマー生成を用いた手法がうまくいかなかったので、今度はxTBを使った2面角のスキャンを試したいと思います。こちらも参考記事2で紹介されていたものです。
xTBはオープンソースのSemiempirical Extended Tight-Binding(半経験的拡張強結合近似(?))プログラミングパッケージです。Wikipedia-強結合近似によると、 「系の波動関数を各原子の場所に位置する孤立原子に対する波動関数の重ね合わせにより近似する手法」で「量子化学で用いられるLCAO法と密接な関係がある」そうです。*9
conda-forgeにあるのでcondaでインストールできます。*10
参考記事2で行われている方法は「RDKitで作成したコンフォマーをベースにしてxTBでエネルギー最適化を行おう!」というものです。
ここでは先に発生させたRDKitのコンフォマーを使います(SetMMFFOopTerm(False)していないもの)。インプットが悪すぎるかな?とは思いますが、今回はコードと使い方の学習重視ということでお許しください。
xTBで計算を行うには以下の点に注意すれば良さそうです。
- 入力ファイルの準備: SDFでOK
- 制約をかけたい2面角の原子のインデックスを設定:xTBは1始まりでRDKitの0始まりとズレるのに注意
- 2面角の制約は
.inpファイルに記載する - 出力ファイルは
xtbopt.sdf
各角度の計算結果の構造を保持したいので、参考記事と少しコードを変えています。
# コード参考元 2; https://pschmidtke.github.io/blog/torsion/dihedral/oss/opensource/rdkit/xtb/energy/2021/02/16/torsion-angle-scans-xtb.html # jupyter notebookから実行するためosを利用する import os # 検証したい2面角の角度 angles=range(0, 180, 10) # エネルギーを格納するリスト xtbenergy=[] # 結果の構造を格納するリスト output_structures = [] # rdkitで生成したコンフォマーを使ってループを回して計算 # xTBの入力に使うsdfを作成 for idx,deg in enumerate(angles): w = Chem.SDWriter('mol_1H.sdf') w.write(model_1H,confId=idx+1) w.close() # 2面角のatom indexを設定。1ずれるので(3, 8, 9, 10)ではなく(4, 9, 10, 11) atoms = '4,9,10,11' # 2面角制約の設定に使うxtb input fileを書く fh = open("dihedral_constraint.inp","w") fh.write("""$constrain force constant=1.00 dihedral: {},{} $end""".format(atoms,float(deg))) fh.close() # xTBの実行 # OMP_STACKSIZEは並列計算のための設定(計算環境に合わせて変更) # OMP_NUM_THREADSは計算に使えるスレッドの数(計算環境に合わせて変更) os.system("export OMP_STACKSIZE=4G && export OMP_NUM_THREADS=4,1 && xtb mol_1H.sdf --opt vtight --charge 0 --input dihedral_constraint.inp") # 出力のxtbopt.sdfに含まれる情報からエネルギーを取り出す。 sdr=Chem.SDMolSupplier("xtbopt.sdf") for xtbmol in sdr: xtbenergy.append(xtbmol.GetProp("total energy / Eh")) # 構造を保持する output_structures.append(xtbmol)
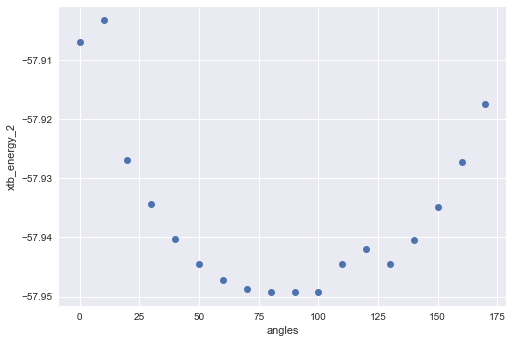
結果のエネルギーをプロットしてみます。xtbenergyリストに格納されているのはstr型なのでfloatに変換しておきます。RDKitの計算結果とは単位が異なる(ハートリーエネルギー, Eh)ことにご留意ください。
xtb_energy = [float(i) for i in xtbenergy] plt.plot(angles, xtb_energy, 'o') plt.xlabel('angles') plt.ylabel('xtb_energy')
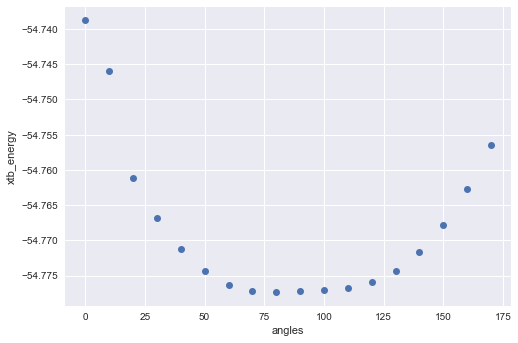
滑らかなプロットになりました。xTBの入力に使ったRDKitの計算結果(一つ目)と見比べると大きな違いです。
構造を眺めてみましょう。
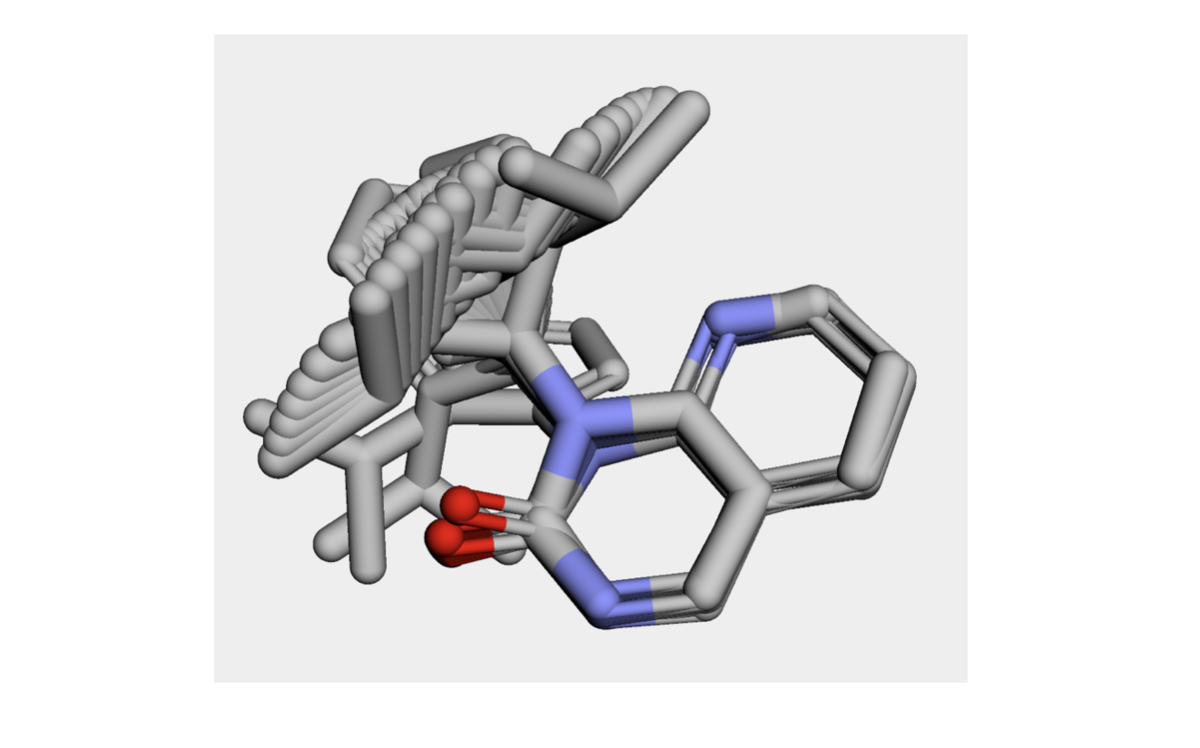
xTBで最適化した構造ではほとんどのコンフォマーで平面性が再現できているようです!!すごい!!計算手法でこんなに変わるんですね!
念のため、最適化に伴って2面角の制約が崩れていないか確認しておきます。
from rdkit.Chem import rdMolTransforms angle_check = [] for m in output_structures: conf = m.GetConformer(0) ang = rdMolTransforms.GetDihedralDeg(conf, 3,8,9,10) angle_check.append(int(ang)) print(angle_check) # [-3, 10, 22, 31, 41, 50, 60, 70, 79, 89, 99, 109, 119, 129, 138, 148, 158, 167]
少々ズレはありますが、10°刻みで0° ~ 170°の18ポイントとる事ができていました。
3. 別のモデル化合物2で検証
RDKitのコンフォマー生成とxTBを組み合わせる事でそれらしい結果が得られる事が分かりました。別のモデル化合物で同じ計算を行うとどうなるか、試してみましょう。
# モデル構造作成 model_2 = Chem.MolFromSmiles("CC(C)c1cccc(C)c1n3c(=O)ncc2cccnc23") # 水素原子付加 model_2H = AllChem.AddHs(model_2) # Atom Indexの追加 for i, a in enumerate(model_2H.GetAtoms()): a.SetAtomMapNum(i) Draw.MolToImage(model_2H)
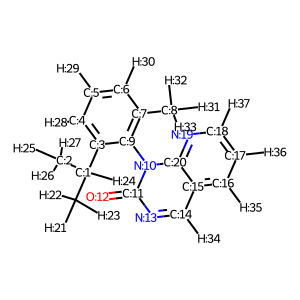
モデル化合物2で目的とする2面角のatom indexは3, 9, 10, 11でした。
xTBに入力するベースのコンフォマーをRDKitで生成します。
# モデル2でコンフォマーを生成 # 初期立体構造の作成 AllChem.EmbedMolecule(model_2H,randomSeed=10) conformer=model_2H.GetConformer(0) m_2 = copy.deepcopy(model_2H) # MMFF力場の設定 m_2_p = AllChem.MMFFGetMoleculeProperties(m_2) energy_2=[] angles=range(0,180,10) confid_2=0 for angle in angles: ff2 = AllChem.MMFFGetMoleculeForceField(m_2, m_2_p) # モデル2に合わせた2面角のatom indexを指定 ff2.MMFFAddTorsionConstraint(3, 9, 10, 11, False, angle - .1, angle + .1, 10000.0) ff2.Minimize() energy_2.append(ff2.CalcEnergy()) confid_2 += 1 new_conf = Chem.Conformer(model_2H.GetNumAtoms()) for i in range(model_2H.GetNumAtoms()): new_conf.SetAtomPosition(i, (m_2.GetConformer(-1).GetAtomPosition(i))) new_conf.SetId(confid_2) model_2H.AddConformer(new_conf)
立体構造とエネルギーはこんな感じでした。
xTBの最適化をおこないます(indexの設定以外は同じです)。
# 検証したい2面角の角度 angles=range(0, 180, 10) # エネルギーを格納するリスト xtbenergy_2 =[] # 結果の構造を格納するリスト output_structures_2 = [] # rdkitで生成したコンフォマーを使ってループを回して計算 # xTBの入力に使うsdfを作成 for idx,deg in enumerate(angles): w = Chem.SDWriter('mol_2H.sdf') w.write(model_2H, confId=idx+1) w.close() # 2面角のatom indexを設定。1ずれるので(3, 9, 10, 11)ではなく(4, 10, 11, 12) atoms = '4, 10,11,12' # 2面角制約の設定に使うxtb input fileを書く fh = open("dihedral_constraint.inp","w") fh.write("""$constrain force constant=1.00 dihedral: {},{} $end""".format(atoms,float(deg))) fh.close() # xTBの実行 # OMP_STACKSIZEは並列計算のための設定(計算環境に合わせて変更してください) # OMP_NUM_THREADSは計算に使えるスレッドの数(計算環境に合わせて変更してください) os.system("export OMP_STACKSIZE=4G && export OMP_NUM_THREADS=4,1 && xtb mol_2H.sdf --opt vtight --charge 0 --input dihedral_constraint.inp") # 出力のxtbopt.sdfに含まれる情報からエネルギーを取り出す。 sdr=Chem.SDMolSupplier("xtbopt.sdf") for xtbmol in sdr: xtbenergy_2.append(xtbmol.GetProp("total energy / Eh")) # 構造を保持する output_structures_2.append(xtbmol)
エネルギーはこんな感じ。
モデル2ではモデル1と比べて置換基を増やしたためか、プロットに谷が2つできました。
立体の確認です。別々のmolオブジェクトとしてリストに格納しているのでAlignMolConformersではなくAlignMolを使っています。
v = py3Dmol.view(width=600, height=600) for mol in output_structures_2: AllChem.AlignMol(prbMol=mol, refMol=model_1H, atomMap=[(i, i) for i in [10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20]]) IPythonConsole.addMolToView(mol, view=v) v.setBackgroundColor('0xeeeeee') v.zoomTo() v.show()
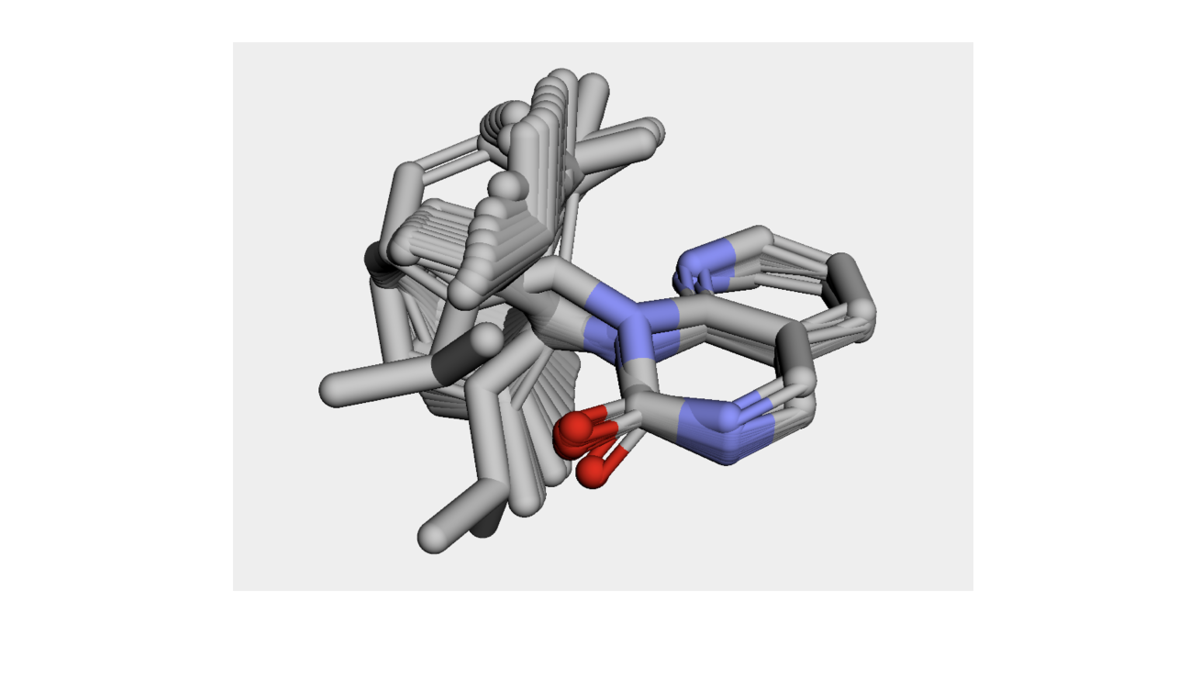
今回もxTBの構造最適化により平面性の崩壊はかなりマシになっているようです。
4. 2つのモデル構造の比較
2つのモデル化合物について2面角とエネルギー変化の計算ができました。合わせてプロットしてみたいと思います。
絶対値での比較はズレが大きいので、それぞれのコンフォマーの最小値との相対的なエネルギー差をとってプロットします。
# それぞれのモデルの最小値と比べた相対的なエネルギーのリスト rel_xtb_energy = [i - min(xtb_energy) for i in xtb_energy] rel_xtb_energy_2 = [i - min(xtb_energy_2) for i in xtb_energy_2] # プロット plt.plot(angles, rel_xtb_energy, label="model_1") plt.plot(angles, rel_xtb_energy_2, label="model_2") plt.xlabel('angles') plt.ylabel('relative_xtb_energy') plt.legend() plt.show
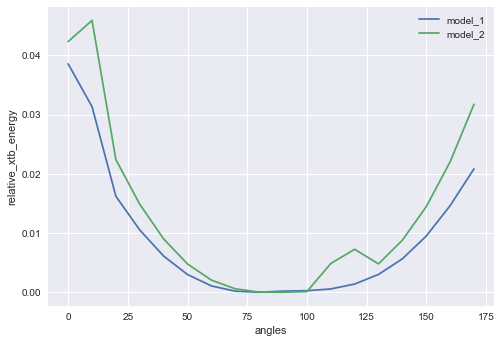
モデル化合物1が青色、モデル化合物2が緑色です。
重ねてみるとどちらも最安定は軸に沿って直交した90°付近にあり、置換基の増えたモデル化合物2は0°、180°付近に近づくに従ってモデル化合物1よりもエネルギーが高く(回転障壁が高く)なっているようです。
それぞれの最大の相対的エネルギー値の差を比較してみましょう。
# 最大の相対的エネルギー値の差 diff_max = max(rel_xtb_energy_2) - max(rel_xtb_energy) print(diff_max, "Eh") # 0.007345450856000468 Eh # 単位を変換(1 hartree = 627.51 kcal/mol) diff_max_kcal = diff_max * 627.51 print(diff_max_kcal, "kcal/mol") # 4.609343866648854 kcal/mol
PC Chem Basics.comさんの記事(1Hartreeは何kcal/mol?(単位換算と物理定数))を基に換算したところ、モデル化合物1と2の最大のエネルギー障壁の差は4.6 kcal/molとなりました。
ところでこれらのモデル化合物はSotorasibの文献中の化合物(18と24)を簡略化したものです。比較するとこんな感じ。
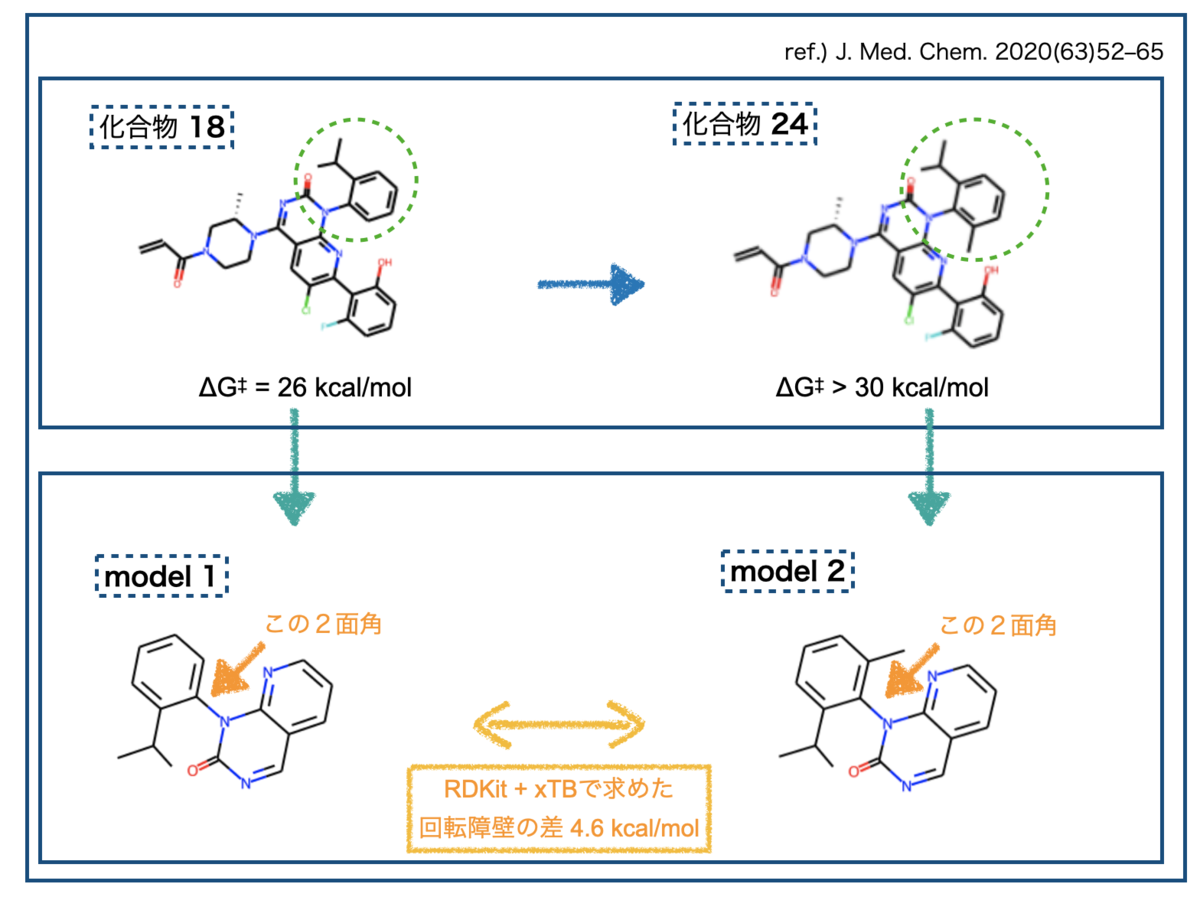
化合物18から24への変換でエネルギー障壁は4 kcal/mol以上上昇したとのことですが、今回のモデル化合物の検証結果は4.6 kcal/molでした。
ひょっとして結構良い結果でてる????
おわりに
以上、今回は医薬品と回転異性体をテーマに、その実例の紹介と回転障壁を題材とした計算を試してみました。
回転異性体は構造式をパッとみてわかる不斉中心が無いので分かりづらいように思います(私だけ??)。こんな時、気軽にちょっと計算してみてエネルギー差がわかったりしたら「あーはいはいそんな可能性もあるのねー」と納得しやすく便利な気がします。
計算の題材としては、近年の話題の医薬品Kras G12C阻害薬Sotorasib(AMG510)文献上の化合物をベースにしてみました。
インターネットの賢い人々の検討結果をコピー&ペーストしたところ、意外にも(?)それらしい結果が得られて正直びっくりでした。素晴らしい情報を公開してくださっている皆様に感謝です。
実際には色々な問題点に目をつぶった、無理矢理な議論をしてしまっています。xTBでマシになったとはいえ平面性が崩れたりしていました。。。
もっと良い方法があるよ!お前のやり方はあまりにもひどい!頭悪すぎ!等々、ご意見いただけると嬉しいです。
なお、本記事はtorsion driveって用語格好いい!と思って書きました。*11
Calculation drives new chemistry!!!
知らんけど
*1:言わずもがな、こちらもノーベル化学賞(2001年)ですね
*2:アレン(allene)みたいなそもそも回転できないものもあります。念のため。
*3:参考: FutureMed.Chem.(2018)10(4),409–422
PuMedでFull textよめます。
*4:もう一つACSのwebinar資料を参考にしています。こちらは何故かインターネット上でPDFが閲覧できるものの出どころ不明なので引用は避けます。
*5:Creative Commonsでは無さそうなので図表の引用は控えます
*6:図中の構造式は論文のSupporting Informationで提供されているSMILESをもとにRDKitで描画しました。
*7:他にはこちらの記事のようにatom indexを確認する方法もあるそうです:RDKitで原子に番号を表示できなくて困っていた話
*8:参考:【RDKit】RWMol で分子(Mol instance) を編集する
*9:誰か説明してください
*10:mambaが使えるならそっちの方が速いかも…
*11:創薬アドベントカレンダーの@iwatobipen先生の記事 Compare torsion drive results between C and O linker of specific structure を読んで、回転って面白いなと思ったのがきっかけです。アドカレに間に合わなくてすみません。