PyMOLをJupyter Notebookと連携させる方法について
前回の記事で、PyMOLのスクリプト機能を利用して3D PSAを求めました。『PyMOL=生物学向けの描画ソフト』と思い込んでいたのですが、まだまだ便利な機能がたくさんあって、化学方面にも応用できそうです。
素敵なPyMOLですが、プログラミングができない私にとってコマンドラインでの操作はとっつきづらいです。コマンドと一緒にコメントや出力結果を残しやすいJupyter notebookで使えた方が後でたどりやすいのになー、と、いうことでJupyterから使う方法を調べました。
PyMOL WikiにJupyterという、そのものな項目があったので記載の方法を試していきたいと思います。ほぼそのままです。
検証する操作の確認
Jupyter notebookを試す前に、検証で行う操作を念のためPyMolアプリのGUIで確認します。
アミノ酸残基はBuildメニューのResidueから選択して作成することができます。コマンドラインでfragment alaとするか、cmd.fragment('ala')としてもOKです。

画像の保存はFileメニューのExport Image Asあるいは右上のDraw/Rayパネルからできます。コマンドラインで行うには、png testとすると作業ディレクトリの中に「test.png」として保存できます。cmd.png('test')でもOKです。

詳しい使い方はPyMol Bookが日本語でわかりやすいです。なお、cmdはPyMOL Python APIを使うためのモジュールで、PyMol上のコマンドラインを使うのであればわざわざ使わなくても良いと思いますが、以降の内容との関係するので併記しました。
JupyterからPyMOL
では本題、Jupyter notebookからPyMOLを利用する方法です。PyMol Wikiには大きく2つ、直接PyMOL APIを使う方法と、サードパーティーのライブラリを使う方法が書かれています。それぞれ制限があるようです。
概要はこんな感じです。
直接PyMOL APIを使う
同じスレッド
まず、同じスレッドで使う方法です。こちらはGUIの画面をみることはできません(headless)。
操作は簡単。Jupyter notebookを起動し、PyMOLのモジュールを読み込むだけです。
# cmd モジュールのインポート from pymol import cmd # アラニンを構築し、拡大した後に画像を保存 cmd.fragment('ala') cmd.orient() cmd.png('./test.png', ray=1)
「PyMOL not running, entering library mode (experimental)」とかいう出力がでてきますが問題なく使えます。PyMOL WikiのLaunching From a Scriptによると、PyMol 2.1から、cmdを使うと自動的にメインスレッドでバックエンドプロセスをGUI無しで起動するらしいので、その関係だと思います。*1
さて、上記で無事「test.png」が作成されたので中身を確認してみます。
from IPython.display import Image Image(filename='./test.png')

うまくいきました!
非同期スレッド
次に、非同期スレッド(asynchronous thread)で使う方法です。こちらはGUIが使えます。
が、残念ながらmacOSではこの操作を行えません(GitHub Issue)。
コマンドだけコピペしておきます。
# PyMOLウィンドウを開く import sys import pymol _stdouterr = sys.stdout, sys.stderr pymol.finish_launching(['pymol', '-q']) sys.stdout, sys.stderr = _stdouterr # PyMOLウィンドウにアラニンを読み込む from pymol import cmd cmd.fragment('ala')
コマンドラインモード
PyMOL Wikiから外れますが、コマンドラインモードであればMacでもJupyterからPyMOLを立ち上げることができます。起動のオプション-cを使うと、GUIなしのコマンドラインのみのモードとなり、バッチプロセスに使うことができます(PyMOLWiki Command Line Options)。
import pymol # PyMolをコマンドラインモードで立ち上げる pymol.pymol_argv = ['pymol','-c'] pymol.finish_launching() # Detected 4 CPU cores. Enabled multithreaded rendering.
ターミナルにPyMOL起動時のバージョン情報が表示され、起動がうまくいったことがわかります。オプション-qをつければこの表示が出ないようにすることもできます(quietモード)。
注意点として、このPyMOLの立ち上げ方はCPUを100%使うらしいです*2。Macが悲鳴をあげるので不要であればcmd.quit()で終了しておいた方が良さそうです。
# コマンドラインモードのPyMOLを終了
cmd.quit()
RPCコミュニケーション
つづいて、Jupyter notebookとは別に立ち上げたPyMol(スタンドアローン)をRPCコミュニケーションで操作する方法です。RPC(Remote procedure call、遠隔手続き呼び出し)は「プログラムから別のアドレス空間にあるサブルーチンや手続きを実行することを可能にする技術」で、「遠隔相互作用の詳細を明示的にコーディングする必要がない」そうです(Wikipedia 遠隔手続き呼び出し)。・・・よくわかりません。
まず、pymol -Rで立ち上げておきます。-RオプションはRPCサーバーを立ち上げるためのオプションです。
pymol -R # xml-rpc server running on host localhost, port 9123
PyMOL GUIアプリが立ち上がりますが、上記のように表示され、xml-rpc serverがlocalhostのポート番号9123で起動していることがわかります。
このRPCサーバーにJupyter notebookからアクセスするにはPythonのxmlrpc.clientモジュールを使えば良いそうです。
# Jupyter notebookからPyMOL にRPCコミュニケーション import xmlrpc.client as xmlrpclib cmd = xmlrpclib.ServerProxy('http://localhost:9123') cmd.fragment('ala')
以上を実行すると、PyMOLのGUIウィンドウにアラニンが読み込まれます。
xmlrpc.client.ServerProxy()でRPCコミュニケーションを管理するためのオブジェクトを生成し、これにPyMOL Python APIを扱うためのモジュールcmdと同じ名前をつけておくことで、APIを扱う感覚で外部のPyMolを操作ができているようです。・・たぶん。
以上が、直接PyMol APIを使う方法です。
サードパーティーライブラリ
つづいてサードパーティーのライブラリを使った方法についてです。ライブラリとしてIPyMOLとRDKitが言及されていますがPyMOL Wikiには詳細は書かれていません。
ライブラリの説明やGitHubをたよりに試してみましょう。
IPyMOL
まずIPyMOLです。IPythonからPyMolのセッションを制御するためのライブラリで、こちらもRPCコミュニケーションを利用します(MITライセンス)。
「Jupyter Notebookできちんと見せたい時や、PyMOLスクリプトのプロトタイプを簡便に作成したい時に理想的なツール」とのことなので、今回の目的にまさにぴったりなライブラリです。
pipでインストール可能(ipymol - PyPI)ですが、古いバージョンらしく不具合があるらしいです(GitHub issue #21)。GitHubバージョンは修正されているらしいので、直接インストールしてみます。
pip install git+https://github.com/cxhernandez/ipymol.git
IPyMOLでは、さきと同様に、あらかじめpymol -Rで立ち上げておいたPyMOLに接続できますが、IPyMolからstart()でPyMOL RPCサーバーを起動することもできます。閉じる時はstop()です。
from ipymol import viewer as pymol pymol.start()
PyMOLのGUIが立ち上がりました!今回は「xml-rpc server running on host localhost, port 9124」と出たので、ポート番号が先とは異なるようです。
アラニンを読み込むには以下のようにします。
pymol.fragment('ala')
display()を使うことでPyMOL GUIのディスプレイをJupyter Notebook上に描画することができます。
pymol.display()

IPyMolで利用可能なAPIは以下を打ち込むと確認できます。出力は多いので省略します。
print(pymol._server.system.listMethods())
上記のリストに含まれていないコマンドを使いたい時はpymol.do("実行したいコマンド")とすればOKです。
使い方の実例はこちらのノートブックが参考になります。
終わったらstop()でウィンドウを閉じられます。
# IPyMOLで立ち上げたPyMOLの終了
pymol.start()
RDKit PyMolモジュール
続いてRDKitからPyMolに RPCコミュニケーションする方法です。Chem.PyMolモジュールを利用すればOKで、化学の新しいカタチさんが「RDKitからPyMOLを利用する」で詳しい使い方をご紹介してくださっています。
参考までにこれまでと同じ処理を確認します。
まず、pymol -RでXML-RPC サーバーを立ち上げておきます。
RDKitで必要なモジュールをインポートし、MolViewerオブジェクトを作成します。
from rdkit import Chem from rdkit.Chem import PyMol v = PyMol.MolViewer()
MolViewerに含まれていない操作を行うには、sever.do("実行したいコマンド")とすれば良いそうです。アラニンフラグメントを読み込み、GetPNG()でJupyter notebook上に描画します。
# アラニンの読み込み v.server.do("fragment ala") # ズーム v.Zoom('ala') # PyMOL GUIの画面をPNGで取得 v.GetPNG()

SaveFile()を使うことで画像として保存することもできます。
折角ですので、ついでに前回の記事で取り上げた3D PSAのスクリプトを実行してみましょう!
まずDeleteAll()で一度全て消した後に、LoadFile()でファイルを読み込みます。「conformer-00.xyz」というxzyファイルを「conf1」という名前のオブジェクトとしてロードしています。
# viewewrのオブジェクトを全て消す v.DeleteAll() # ファイルの読み込み v.LoadFile('./conformer-00.xyz', 'conf1')
別途作成したスクリプト(psa3d.py)をつかえるように読み込み(run)、計算を実行します。
# スクリプトの実行 v.server.do('run ./psa3d.py') # 計算の実行 v.server.do('set solvent_radius=1.4; set dot_density=4; set dot_solvent=1; psa3d')
無事計算が実行され、PyMOL上にSA 3D PSAの値「201.973」が表示されました。
以上、サードパーティライブラリの動作も確認できました。
まとめ
以上、PyMOLをJupyter notebookで利用するための方法の紹介でした。ほとんどPyMOL Wikiそのままですが。。。
PyMOLのスクリプトは色々な方が作成、公開してくださっているようなので、Jupyter notebookとうまく連携させればプログラミング苦手な私でももっと活用できるかもしれません。
Jupyter notebookで操作して出力された結果を、PyMOLから受け取ってJupyter Notebookに表示することもしたかったのですが、やり方がよくわかりませんでした。サブプロセス(?)を使えば良いのかもしれませんが私のキャパシティでは無理でした。。。
ではでは。
*1:どういうことかはさっぱりわかりません。。。
PyMolで3D PSAを計算しようとする話
先日の記事で、下記文献を例に3次元構造の指標であるRadius of gyrationについて取り上げました。
論文1) Solution Conformations Shed Light on PROTAC Cell Permeability
Yoseph Atilaw, Vasanthanathan Poongavanam, Caroline Svensson Nilsson, Duy Nguyen, Anja Giese, Daniel Meibom, Mate Erdelyi, and Jan Kihlberg
ACS Medicinal Chemistry Letters Article ASAP DOI: 10.1021/acsmedchemlett.0c00556
https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00556 (CC-BY 4.0)
今回は同文献で使われていた別の指標SA 3D PSAについて調べました。
合わせて、この記事では以下の内容に触れています。
- シェルスクリプトによるテキストファイルの処理
- RDKitに適したxyz形式の変換方法
- PyMolのコマンドと自作関数の使い方
SA 3D PSAについて
SA 3D PSAの定義や求め方について上記文献には記載がありませんでした。求め方として著者らのグループ(Uppsala大学、 Jan Kihlberg教授)が2020年に発表した下記文献が引用されていましたが、オープンアクセスではなかったため中身を確認できませんでした。
論文2) Solution Conformations Explain the Chameleonic Behaviour of Macrocyclic Drugs
E. Danelius, V. Poongavanam, S. Peintner, L. H. E. Wieske, M. Erdélyi, J. Kihlberg
Chem. Eur. J. 2020, 26, 5231.
さらにたどると同グループの以下の文献にあたりました。
論文3) Impact of Dynamically Exposed Polarity on Permeability and Solubility of Chameleonic Drugs Beyond the Rule of 5
Matteo Rossi Sebastiano, Bradley C. Doak, Maria Backlund, Vasanthanathan Poongavanam, Björn Over, Giuseppe Ermondi, Giulia Caron, Pär Matsson, and Jan Kihlberg
Journal of Medicinal Chemistry 2018 61 (9), 4189-4202
こちらも論文の中身自体は確認できませんでしたが、Supporting Informationに3D PSA calcuration methodsという記載がありました。
ざっと以下のような内容です。
- 3Dで分子の極性原子(polar atom)の占める面積
- 分子の表面積(molecular surface area)は、プローブの半径を0Åとしたもの
→ 3D PSA - 溶媒接触表面積(solvent accessible surface area)は、プローブの半径を1.4Åとしたもの
→ SA 3D PSA
これら2種の3D PSAの計算にはPyMol 1.7.0.1が使われたとのことです。
また、極性原子は
と、なっています。このうち4. 部分電荷はPM3 semi-empirical methodで計算した値で、SIから判断する限り閾値は0.6のようです。
なお、こちらのJ. Med. Chem.文献(論文3)よりも後に出版された、先のChem. Eur. J.文献(論文2)は、電荷の計算方法が改善しており、その手法をACS. Med. Chem. Lett.文献(論文1)では採用しているようです。
ざっとですが3D PSAの概要がつかめました。
同様な値がオープンソースツールを使って計算できるか?早速やって見ましょう!
入力構造の準備(シェルスクリプトの練習)
先の記事で、論文1 のSupporting Informationからコンフォマーの配座情報を取り出しました。このSIには全部で24個のコンフォマー情報が含まれています。
今回はテキストファイル処理の練習も兼ねて、すべて取り出して見たいと思います。・・・シェルスクリプト苦手。
処理対象のファイルは前回pdfminer.sixでPDFから取り出したテキストのうち、コンフォマー情報を含むテキストファイルです(SI.txt)。
sedコマンドを使えば良いとのことですが、MacのsedはBSD版というもので、Linuxのsed(GNU版)と挙動が異なるようです。
後者の方が使い勝手が良いらしいので、GNU版sedをインストールしてみます。*1
Homebrewで簡単に入れられます。
brew install gun-sed
注意点として、sedの代わりにgsedとコマンドを入力する、とのこと。
ここで、行いたい処理は下図の通り、余分なテキストの削除とxyz形式に合わせるためのテキスト追加です。

ファイルを上書きするオプション-iと、文字列の正規表現を使って、以下のようにしました。*2
(Jupyter notebookで行なっているのでマジックコマンドを含めて!gsedとしています)
# 改ページの文字列(\f)の削除 !gsed -i 's/\f//g' SI.txt # ページ番号の行を削除。行の先頭が数字となるもの(「^[0-9]」)。 !gsed -i '/^[0-9]/d' SI.txt # 空行を削除。「^ *$」で改行のみおよび空白のみを指定。 !gsed -i '/^ *$/d' SI.txt # conformer_xx行の前に、原子数(131)の行を追加。行の先頭がconとなるもの(「^con」)。 !gsed -i '/^con/i 131' SI.txt
上記で得られたファイル(SI.txt)の中身は、24個のコンフォマーの情報が連続する3192行のファイルです。
余談ですが、一番最初の「改ページのメタ文字(\f)」を削除しなければいけないことに気づくまでに2時間くらいかかりました。。。。
何度gsedを実行しても、テキストエディタを変えて確認してもうまくいかない理由がわからず、結局「Pages」で開いて改ページが含まれているに気づきました。*3
皆様もPDFから起こしたテキストにはくれぐれもご注意を!
さて、気を取り直して!
コンフォマー1つあたり133行なので、stripコマンドを133行ごとに別のファイルに区切ります。
ここでもMacでGNU版のstripを使うため、Homebrewでcoreutilsをインストールし、gstripコマンドを使います。
brew install coreutils
分割後のファイルは「conformer-数字.txt」となるようにします。
!gsplit -l 133 -d SI.txt conformer-
「conformer-XX(Xは数字)」というファイルが「XX = 00 ~ 23」の24個できました。
拡張子をxyzに変更するためrenameでファイル名を変更します。これもインストールが必要のようです。
brew install rename
-aオプションで末尾に「.xyz」を追加します。
!rename -a .xyz conformer-*
できました! 拡張子が付与され自動的にAvogadroに紐づけられました。

XYZ2molでRDKit Molオブジェクトに一発変換
先の記事ではxyzファイルの変換にOpenBabelを使いましたが、出力構造に難がありマニュアルで修正する必要がありました。
今回はxyz2mol(GitHub)を使ってみたいと思います。
こちらはRSCのワークショップ(open-source-tools-for-chemistry)で紹介されていました。こちらに関連資料があります。
xyz2molの使用には以下が必要とのことなので、インストールします。
- rdkit (>= 2019.9)
- networkx
conda install -c anaconda networkx
続いて、githubから作業フォルダにそのままcloneします。
!git clone https://github.com/jensengroup/xyz2mol
READMEには「フォルダを移動してmakeするように」、とかいてありますが、しなくてもimportできました。
import xyz2mol.xyz2mol as xyz2mol
先のワークショップのうち、「Chemistry in the cloud: leveraging Google Colab for quantum chemistry (by Jan Jensen氏)」のGoogle Colab Notebook (URL→ https://bit.ly/37fIYbp ) に利用例があるので、コードをお借りして進めます。
まず、read_xyz_file()でxyzファイルを読み込みます。原子(atom)、電荷(chage)、xyz座標(coordinate)を返すので、それらをxyz2mol()に渡します。するとRDKitのMol objectを含むリストが得られます。
atoms, charge, xyz_coordinates = xyz2mol.read_xyz_file('conformer-00.xyz') conf_mol_0 = xyz2mol.xyz2mol(atoms, xyz_coordinates, charge=charge) print(type(conf_mol_0)) print(len(conf_mol_0)) print(type(conf_mol_0[0])) # <class 'list'> # 1 # <class 'rdkit.Chem.rdchem.Mol'>
無事RDKitのMolオブジェクトが得られました!
少し時間がかかる印象ですが、OpenBabelと異なり一発でxyzファイルから生成できるのは便利ですね。
肝心の構造はどうでしょうか?py3Dmolで描画し、OpenBabelでMolファイル変換した構造と比較してみました

無事正しい構造が起こせていそうです!!これは便利!
ぶっちゃけxyzファイルを変換しなくても以降の計算は可能なのですが、ファイル変換の方法なんてなんぼあっても良いですからね!
3D PSAの計算
さて、構造の準備と変換方法が分かったので、目的の3D PSA計算に挑戦したいと思います。
冒頭確認した通り、必要な情報は
- 各原子のxyz座標
- 各原子が極性原子に該当するか(原子タイプと部分電荷)?
でした。1.は手元にあるので、2.を求める方法があるか検討してみました。
部分電荷の計算に失敗
WinMopac
論文3) J. Med. Chem. 2018の記載に従えば、PM3 semi-empirical methodで部分電荷を求める必要があります。
PM3って何でしょう???量子力学さっぱりわかりません。
PC CHEM BASICS.COMさんの記事(MOPACの入手とインストール方法)によれば、とりあえずMOPACがお手軽だそうです。
そこで記載を参考に、Macで実行するため以下をダウンロードしました。
ネット回線が弱いのですごい時間かかりました。。。。
で、いざWinPopacに構造を投げ込んだところ「最大で処理可能な原子数を超えてます」みたいエラーで動きませんでした。
残念。お正月休みを返して欲しい。
xTB
ついで、xTB(extended tight binding)を利用してみることにしました。
xTBはオープンソースで利用可能で、さきにご紹介したRSCのワークショップで使われていたソフトウェアです。
検索した結果、「半経験量子計算法:Tight-Binding(強結合近似)計算」という言葉がでてきました。さっぱりわかりません。。。
とりあえずPM3もsemi-empricalって言ってるしこれを試してみよう!ということでインストールしました。
condaで入ります。
conda install -c conda-forge xtb
xTBのドキュメントによると、single point calculationを行うにはシェルでxtb coord --sccとするだけのようです。
coordは原子座標でxyzファイルを入力として使うこともできます。また、--sccはself-consistent charge calculationの略のようです。
以下を実行しました。
!xtb ./conformer-00.xyz --scc
・・・止まりました。
今回も!何の成果も得られませんでした!!!!
あきらめました。
座標のみで計算
諦めが早いので、部分電荷にこだわらず座標と原子タイプ(N、O、NH、OH)の情報のみで計算することとしました。
論文3) J. Med. Chem. 2018のSupporting InformationよりPyMolで求められるようなので、こちらの情報に従って試していきましょう。
PyMolで使用するスクリプトの準備
計算用のスクリプトを用意し、PyMolのコマンドラインから使用するだけらしく、スクリプトの情報もありました。
まず、以下を記載したファイルpsa3d.pyを用意します。
from pymol import cmd def psa3d (arg1=0.6, arg2=-0.6): obj_list = cmd.get_names('objects') for obj in obj_list: cmd.select('noh', '(pc.>'+str(arg1)+'|pc.<'+str(arg2)+'|(e. n OR e. o OR e. h AND(neighbor e. n OR e. o))) AND ' + obj) print("3DPSA_noh06 %12s %.3f" % (obj, cmd.get_area('noh'))) return obj cmd.extend("psa3d", psa3d)
このスクリプトでは大きく3つのことが行われています。
1.でimportするcmdモジュールですが、これによりPython Python APIにアクセスすることができ、cmd.<command-name>(argument, ...)という使い方ができます。*4
3.cmd.extend(文字列, 関数)は、PyMol外部の関数をPyMolスクリプトで使うために、新しい名前(文字列)のコマンドに結びつけます。ここでは自作の関数psa3d()の名前をpsa3dと結びつけてるので、PyMolのコマンドライン上でpsa3dとすれば使えるようになります。
それでは関数psa3d (arg1=0.6, arg2=-0.6)の中身をみていきます。
ここで使われているPyMol特有のコマンドは以下の3つです。*5
| コマンド | 説明 |
|---|---|
cmd.get_names('objects') |
PyMol上のオブジェクトの名前を取得(リスト形式) |
cmd.select('name', 'selection') |
'selection'で選択した原子(群)に、名前('name')をつけて、その名前で扱えるようにする |
get_area('selection') |
'selection'で選択された原子(群)の表面積(Å2)を計算 |
これを踏まえると、psa3d()は「PyMolに読み込まれている構造(オブジェクト)について、それぞれ3D PSA算出の対象タイプとなる極性原子を選択し、その原子(群)の表面積を求める」という関数になりそうです。
cmd.select()のselectionがややこしいので、もう少しみてみます。
論文3のSIより、現在考えたい極性原子は以下です。(再掲)
以上をふまえてpsa3d(arg1=0.6, arg2=-0.6)の引数の値を代入してselectionを書き直してみます。
(pc.> 0.6 | pc.<-0.6 | (e. n OR e. o OR e. h AND(neighbor e. n OR e. o))) AND オブジェクト)
上記のうち、「縦線(|)」と「OR」は「または」、「AND」は「且つ」と読み替えれば論理式のように読むことができそうです。
以下のSelection Algebraをふまえると、selectionが目的の原子の選択に対応していることがわかると思います。*6
| Selection Algebra | 説明 |
|---|---|
pc. |
partial_charge(部分電荷)「pc. <演算子(ex. 大・小・イコール)> <値>」 |
e. |
elemment(元素記号)「e. <元素記号(ex. 窒素 n、酸素 o、水素 h)>」 |
neighbor |
直接結合する近接原子 |
以上のような条件設定で選んだselectionに対して「noh」という名前を与えて新しいselectionにしているようです。
これでpsa3d.pyの説明は終わりです。・・・思ったより長くなった。
PyMolでスクリプトを利用して計算
用意したスクリプト(psa3d.py)をPyMolで使うには、PyMol起動後、PyMol コマンドラインで以下を打ち込むだけでOKです。
run ~/Desktop/psa3d.py
Pythonファイルのパスは適宜変更してください。今回はわかりやすくするためデスクトップに置いておきました。
次に、計算したい構造ファイルをPyMolに読み込みます。今回は、以前の記事で作成したMolファイルを使いました。
これで準備は完了です!それでは3D PSAを計算しましょう!!
以下のコマンドをコピペすれば2種類の3D PSAをそれぞれ計算することができます。
- 分子の表面積(M 3D PSA) 「
set dot_density=4; set dot_solvent=0; psa3d」 - 溶媒接触表面積(SA 3D PSA)「
set solvent_radius=1.4; set dot_density=4; set dot_solvent=1; psa3d」
コマンドの中身を簡単に説明します。
どちらも最後はpsa3dとなっています。psa3d.pyの最後の行でcmd.extend()を使って、関数psa3d()の名前をpsa3dと結びつけてるので、
PyMolのコマンドライン上でもpsa3dでと打てば関数を使うことができるようになっています。
それよりも前の記述は全て「表面積を求めるコマンド(get_area)」の条件を設定(set)しています。*7
| 設定条件 | 説明 |
|---|---|
dot_solvent |
0 :分子の表面積(molecular surface)を計算(デフォルト) 1 : 溶媒接触表面積(SASA)を計算 |
dot_density |
1 ~ 4:サンプリング密度(density)。高密度(数字が大きい)ほど精度は高いが、処理に時間がかかる |
solvent_radius |
溶媒の半径(デフォルトは1.4) |
上記とコマンドを見比べると、どちらも最も精度の高いdot_density=4を選択していることがわかります。
また、SA 3D PSAではプローブの半径を1.4Åとしていたので、dot_solvent=1とした上でset solvent_radius=1.4という設定が入っています。
これらの設定が関数psa3dの中のcmd.get_area()に反映されているようですね。
さて、コマンドの中身がわかったので、早速計算を実行してみましょう! コンフォマー1の3D PSAはどのような値を取っているでしょうか?
計算自体は一瞬で終わりました。結果はこんな感じです。

それぞれ 213(Å2)、202(Å2)となりました。SA 3D PSAの方が小さくなるのは意外でした。
少し見辛いですが、上図の通り、「酸素原子: 赤色のドット」、「窒素原子: 青色のドット」で極性原子の表面積が表されています。
ではこの計算値、論文1)の文献の値とくらべてどうでしょうか? Figure. 5 を引用します。

Conformer 1のSA 3D PSAは202(Å2)!!同等な値が出ました!今回は部分電荷、あまり関係なかった感じでしょうか?
なお、入力ファイルをmolファイルではなく、座標のみのxyz形式としても同じ結果がでました。
電荷を考えないのであれば3D PSAの計算は原子の座標のみで良さそうです。
芳香属性とか結合の情報はあまり重視しない指標なのでしょうか???
またFig. 5に記載されている残りの2つについても、コンフォマー2のSA 3D PSAは 182.137、コンフォマー3のSA 3D PSAは 194.195と、と同等の結果が得られました。
まとめ
以上、今回は立体構造の指標3D PSAについて調べてみました。論文の本文自体を読めておらず、Supporting Informationの情報のみからの推察ですが、計算結果としてはおおよそ近い値が得られました。
スクリプトが提供されていたお陰ですが、PyMolでさっと計算できてしまうのが個人的にはびっくりでした。描画だけでも盛りだくさんなのに、こんな便利な使い方もできるとは、、、PyMolすごい!
感動したのでPyMolコマンドを調べていたら無駄に長くなってしまいました・・・。
しかし、量子力学がさっぱりわからないです。DFTはおろかHFもよくわかっていません。いったいみなさんどこで勉強されたんでしょうか??
シェルスクリプトの段階で躓いているので、まだまだ道は遠そうです。今回も間違いが多そうなのでご指摘いただければ幸いです。
ではでは!!
*2:参考1 置換・行や文字の削除などの文字列操作 (sed)
参考2 sedコマンドで覚えておきたい使い方12個(+3個)
*4:PyMolのコマンドリファレンス(古い?) http://pymol.sourceforge.net/oldman/S1000comref.html
*5:コマンドの説明は間違っているかもしれません。すみません。
*6:PyMol Wiki Selection Algebra
RDKitで変換可能なペプチド配列と核酸配列について
以前、マクロ分子の表現HELMについて取り上げた記事の中で、ChEMBLに登録されているモノマー表記を調べました*1。その結果、天然のアミノ酸だけでなく様々な修飾アミノ酸が登録されており、略号(ID)で表せるようになっていることがわかりました。
ところでRDKitも同様に略号を使って構造を表すことができます。@iwatobipen先生のこちらの記事に詳しいです。
とても便利な機能ですが、どのような略号が使えるかわからないと、うまく活用できません。そこで今回はRDKitで扱える略号はどのようなものがあるのか調べてみました。
RDKitでの略号からの構造生成例
まずはイメージを掴むために実際に動きを確認してみましょう。
RDKitのドキュメンテーションのMolFromHELM()の項目では、「Construct a molecule from a HELM string (currently only supports peptides).」となっています。一方でGitHubのissueで指摘されているように、実際にはペプチド以外にも核酸の配列からMolオブジェクトを作成することができます。
確認してみます。
from rdkit import rdBase, Chem from rdkit.Chem import AllChem, Draw from rdkit.Chem.Draw import IPythonConsole, rdMolDraw2D from IPython.display import SVG print(rdBase.rdkitVersion) # 2020.03.2 # ペプチドの例 Glutathione = Chem.MolFromHELM("PEPTIDE1{E.C.G}$$$$")

# RNAの例 RNA_AU = Chem.MolFromHELM("RNA1{P.R(A)P.R(U)P}$$$$")

# DNAの例 DNA_ACGA = Chem.MolFromHELM("RNA1{[dR](A)P.[dR](C)P.[dR](G)P.[dR](A)}$$$$")

確かに、アミノ酸の一文字表記に加えてRNA、DNAの核酸の略号にも対応しているようです。
以上はHELMからMolオブジェクトを生成する例です。
ついで、配列(Sequence)からMolオブジェクトを生成する例も確認しておきます。こちらは上でも引用させていただいた@iwatobipen先生の記事で扱われている機能です。
Plan! Do! Check! Act!
# 描画の設定を変更 IPythonConsole.drawOptions.addStereoAnnotation = True IPythonConsole.drawOptions.annotationFontScale = 1.3 # SequenceからMolオブジェクトを生成 PDCA = Chem.MolFromSequence('PDCA')

できました!*2
flavor=1という引数を与えるとD-アミノ酸も扱えるようになり、小文字でD体を表せます。*3
# D-アミノ酸の使用例 d_PDCA = Chem.MolFromSequence('pdca', flavor=1)

くさび表記は見づらいですが、ステレオ表記から立体が反転していることが確認できます。
コードから利用可能な略号を探ろう!
RDKitドキュメンテーションの情報も最新ではない、ということなので、該当と思われる箇所のコードを確認してみましょう。以下がそれっぽい箇所です。*4
- rdkit/Code/GraphMol/FileParsers/SequenceParsers.cpp
- rdkit/Code/GraphMol/FileParsers/SequenceWriters.cpp
どうやらC++の名前空間として略号とその表す構造が定義されているようです。
さっぱりわかりません。。。
RDKit UGM_2015 Roger Sayle氏の発表資料*5の記述が、仕組みを理解する上で役に立ちました。
- シークエンス情報は、内部でPDBの残基コード(residue number)を使ってエンコードされている
- (residue number のような3文字表記は、)1文字表記よりもたくさんの構造を表現できる
これを踏まえてコード(SequenceParsers)を見直すと、確かにPDBで見られる残基、原子の表現を用いながらオブジェクトを作っている感じが伝わってきます。
ここまでHELMとシークエンスの話をごちゃ混ぜに進めてきました。見通しをよくするために、まずはシークエンスからの構造生成に焦点をあてて見ていきたいと思います。
アミノ酸の構築方法
シークエンスを構造に起こすにあたって、1文字表記から各アミノ酸をどうやって構造におこしているのでしょうか?
SequenceParsers 84行目から抜粋します。
具体的な構造構築の情報は下図の真ん中あたりからです。最初のアラニン(ALA)、アルギニン(ARG)の例を見比べていただくと分かりやすいです。

上図の通り、アミノ酸は以下のように構築しています。
- アミノ酸主鎖を構築(
CreateAABackbone) - 側鎖を構築(
CreateAAAtomで原子を指定し、CreateAABondで結合関係と結合次数を指定)
側鎖の原子の表現にはPDBファイルでよく見かけるγ炭素(CG)、δ炭素(CD)といった表現が使われています。
注意点としては、ALAの表現から分かるようにβ炭素までCreateAABackboneで構築しています。プログラム実装上の都合と思いますが、通常の主鎖とはズレています。
実際に、グリシン(GLY)は以下のようにCreateAABackboneを使わずに表現されています。

シークエンスから構築したMolオブジェクトには上で見た原子の情報が含まれています。AtomオブジェクトのGetPDBResidueInfoというメソッドを利用することで取り出せそうです。
# 一文字表記からアルギニンを構築 arg_mol = Chem.MolFromSequence("R") # 原子の名称を取得しリスト化 atom_names_list = [] for a in arg_mol.GetAtoms(): a_name = a.GetPDBResidueInfo().GetName() atom_names_list.append(a_name) # 原子名と一緒に描画 view = rdMolDraw2D.MolDraw2DSVG(300,300) tm = rdMolDraw2D.PrepareMolForDrawing(arg_mol) # optionで描画にatom labelを追加 option = view.drawOptions() for i, an in enumerate(atom_names_list): option.atomLabels[i] = an option.padding=0.01 option.legendFontSize=20 view.DrawMolecule(tm, legend="arginine with atom labels") view.FinishDrawing() svg = view.GetDrawingText() SVG(svg)

できました!
C末端のカルボキシル基酸素原子はOXTなんですね。なるほど。。
登録されているアミノ酸
構造構築の裏側がわかってきました。デフォルトではどのようなアミノ酸を含んでいるのでしょうか?
コードにある3文字と1文字略号を表にしてみました。

約50種のアミノ酸情報がありました。
標準アミノ酸のL体、D体の他にもセレノメチオニンやオルニチンといったものもあります。
D体の1文字表記はL体の表記を小文字にしたものとなっています。
コードに不備があるのか、D-セリン、D-チロシンは構造生成のための情報が欠けており、利用することができません。
1文字略号sやyでMolオブジェクトは生成されますが、中身は空です。
d_SER = Chem.MolFromSequence('s', flavor=1) print(type(d_ser)) # rdkit.Chem.rdchem.Mol print([a for a in d_ser.GetAtoms()]) # []
登録済みのヌクレオチド
こちらは種類が少ないので簡単に。
塩基として標準的なA、C、G、T、Uが用意されています。
詳細はコードの中身を見ていただきたいですが、どうやらリン酸-糖を別に用意し、略号で指定された塩基を結合させてそれぞれのヌクレオチドを構築しているようです。
他の特徴として、末端の5'側、3'側、それぞれについてリン酸でキャップした構造(PCap5、PCap3)を生成するための情報も組み込まれています。
DNAとRNAのシークエンスの区別は引数flavorで可能です。
シークエンスの種類を区別するflavor
ここまでRDKitで利用可能な略号を確認してきましたが、アミノ酸とヌクレオチドで同じ略号があるため、区別する必要があります。
MolFromSequenceでは、引数flavorの値により、シークエンスがペプチド、DNA、RNAいずれかを区別することができます。

同じシークエンスを使って違いを確認しましょう。Proteinについては既にみたのでRNA、DNAを試します。
# 5'のみリン酸キャップしたRNA RNA_test = Chem.MolFromSequence('ACG', flavor=3)

# 3'のみリン酸キャップしたDNA DNA_test = Chem.MolFromSequence('ACG', flavor=8)

図の通り、シークエンスをヌクレオチドとして認識し、それぞれリン酸キャップと糖の違いが反映されたオブジェクトが生成できました。
以上でMolFromSequence()で構築可能な構造の範囲がおおよそわかりました。
先に見た登録済みのアミノ酸の表を再度見直すと、非標準のアミノ酸は3文字表記のみで、1文字表記がありません。ですが、MolFromSequnece()は1文字表記の配列が対象なので、これらのアミノ酸を利用できません。
これらのアミノ酸はHELMであれば利用可能です。では続いてHELMからの構造生成を見てみましょう!
HELMで利用可能な略号
HELMではモノマーが複数文字の場合、「角括弧[ ]」で囲むことで一つとして扱えます。したがってシークエンスで扱えなかった3文字表記のアミノ酸も利用可能です。ただし略記(ID)が若干異なるので注意してください。
以下の表にまとめました。L-アミノ酸はシークエンスと同様、一般的な1文字略号でOKなので省略しています。

末端のキャップを含めて、非標準のアミノ酸を試してみましょう!
peptide_test = Chem.MolFromHELM("PEPTIDE1{[ac].[Orn].[Glp].[seC].[am]}$$$$")

「N末端: アセチルーオルニチンーL-ピログルタミン酸ーL-セレノメチオニンーNH2:C末端」という構造が生成されました!
HELMは表現の幅を広げやすいのが良いですね!
ヌクレオチドについては冒頭に見た通り、糖について
- RNA : リボース「
R」 - DNA : デオキシリボース「
dR」
を使うことに注意すればOKです。
以上で、RDKitに組み込まれている略号は終了です。
メッセージをペプチドにのせて!
さて、RDKitで扱える配列を眺めてきました。短いエントリですがまとめに代えて。。。
2020年は本当に色々なことがありました。来年のことを言うと鬼が笑う、なんて言いますが、最後はこのメッセージを構造式にしてしまいましょう!
# シークエンスをペプチドに変換 HNY = Chem.MolFromSequence("HAPPYNEWYEAR") # 描画の設定 view = rdMolDraw2D.MolDraw2DSVG(400,400) tm = rdMolDraw2D.PrepareMolForDrawing(HNY) # optionで調整 option = view.drawOptions() option.legendFontSize=20 view.DrawMolecule(tm, legend="Happy New Year") view.FinishDrawing() svg = view.GetDrawingText() SVG(svg)

良いお年を!
*2:サイクルは回ってませんが
*3:RDKit ドキュメンテーション MolFromSequence
*4:Pistoia Alliance Public Resources HELM Parsers and Writersの記述を参考
*5:p21
コンフォメーションとRadius of gyrationによる評価について
巨大なPROTACがなぜ細胞膜を透過するのか?溶液中におけるコンフォメーションの違いから解析した興味深い文献を読みました。
Solution Conformations Shed Light on PROTAC Cell Permeability
Yoseph Atilaw, Vasanthanathan Poongavanam, Caroline Svensson Nilsson, Duy Nguyen, Anja Giese, Daniel Meibom, Mate Erdelyi, and Jan Kihlberg ACS Medicinal Chemistry Letters Article ASAP DOI: 10.1021/acsmedchemlett.0c00556 (https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00556)
CC-BY 4.0の元で公開されています。
この論文中で出てきた Radius of gyration (Rgyr) というものがよくわからなかったので調べてみました。
合わせて、この記事では以下の内容を扱っています。
- PDFファイルからのテキスト情報抽出
- 化学情報のファイル形式xyz形式とその変換
- 論文の概要
- Radius of gyration (Rgyr)
- PDFから座標を抜き出す
- 座標のXYZ形式とPyMolでの確認
- Open Babelによるファイル形式の変換と修正
- RDKitによるRadius of gyrationの計算
- PyMolでradius of gyrationを描画
- まとめ
論文の概要
まずは論文の中身についてざっと紹介します。
PROTACは概して分子量が大きく、LipinskiのRule of 5からは大きく外れた構造をとっています。このような巨大な分子が細胞膜を透過する、経口投与で薬効を示す、というのは非常に驚きです。
では何故膜透過が可能なのでしょうか?
その一つの解として、こちらの論文では、溶液中でのコンフォメーションの違いが寄与しているのではないか?と示唆しています。つまり細胞膜中のような疎水性環境下では、丸まったコンフォメーションをとっており、実質的な体積(大きさ)は小さくなっているのではないか?、ということのようです。
解析されている構造例は以下。グラフィカルアブストラクトがわかりやすいですね。
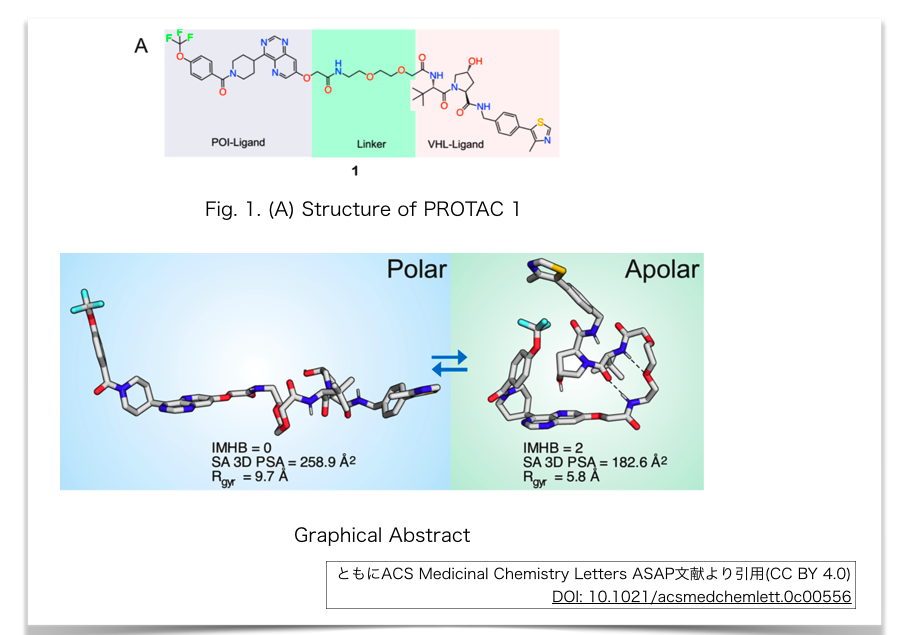
以下の解析手法でコンフォメーションを求めているようです。
- コンフォメーション解析の元となる実験データはNMR
- NMRデータ解析手法はNAMFIS(NMR analysis of molecular flexibility in solution)アルゴリズム
- NAMFISでは様々なコンフォメーションの時間平均であるNMRデータを、各コンフォーメションの占有率に分解して解析可能 *1
- NAMFISに必要なコンフォメーションのアンサンブルはMCMM (Monte Carlo molecular mechanics) conformational searchで生成*2
得られたコンフォメーションを評価する指標として、以下を用いています。
SA 3D PSAの具体的な計算方法は理解できていませんが、化学の新しいカタチさんの記事「トポロジカル極性表面積は計算コストの低い推定PSA」が参考になると思います。
PSAはオクタノール/水分配比率(logP)などと同様に、分子の極性・脂溶性を示す記述子になります。PSAと膜透過性に関する実験データとの間にはよい相関があることが判明しています。例えばあまりに大きなPSA(140 Å2以上)を有する化合物は膜透過性が低いことが経験的に知られています。(化学の新しいカタチ 上記記事より引用)
得られたコンフォメーション解析結果から、PROTAC 1は疎水性環境中では丸まった構造をとっていることが示唆されました。
このコンフォメーションを安定化させる構造的特徴として以下が指摘されています。
- 分子内水素結合(IMHB、intramolecular hydrogen bond)
- 芳香族へテロ環(チアゾール、ピリミジン)間のπ-π interaction
- ヒドロキシ基とピリミジン間の非古典的水素結合(nonclasical hydrogen bond)
IMHBやnonclasical hydrogen bondは、アミドNHやヒドロキシ基を溶媒接触面からマスクすることでもSA 3D PSAを低下させ、膜透過の向上に関与しているのではないか?ということが指摘されています。
この解析結果が非常に興味深いと思ったのは、一見すると2つの大事なユニットをつなぐだけに見えるflexibleなPEGリンカーが、分子内水素結合という形で3次元構造の安定化に関わり、膜透過の向上という物性変化に積極的に寄与している可能性が示唆されている点です。
回転可能な結合数(Nubmer of rotatablebond、NRotB)は極性表面積(PSA)と同様に、一般に膜透過と逆相関することが指摘されている指標のように思います。NRotBを増加させる自由なPEGリンカーが、実は膜透過に大きな役割を果たしているかもしれない、カメレオン的特性のキモ、というのはとても面白い示唆だと思います。
Radius of gyration (Rgyr)
で本題、Radius of gyrationとは何なのか?
Wikipediaによると断面回転半径と訳されるらしく、断面の性質を表すパラメータの一つだそうです。

何のこっちゃ????
英語版のWikipediaの方がもう少しわかりやすいです(Radius of gyration (wikipedia))。
曰く、「慣性モーメント(moment of inertia)を持つ点までの半径方向距離(radial distance)」です、と。
数学的には「重心、あるいは与えられた軸からの物体の各パーツまでの距離の二乗平均平方根」となるそうです。
・・・なるほどわからん。ポリマー科学におけるIUPACの定義も記載されているので、ポリマー界では一般的に使われる指標なのでしょうか???
Wikipediaの表式は軸(axis)についてのものっぽいです。重心(center of mass)についての式はこちらのページにありました。

Rgyrが小さいほど原子が重心に近く、大きいほど遠くなるそうです。つまりRgyrが小さい方が3次元的に小さい(丸まっている?)ということのようです。
とりあえず座標と質量があれば計算できるようです。
ほな、やってみましょうか!!
PDFから座標を抜き出す
先のACS Med. Chem. Lett.論文ではコンフォマーの座標がSupporting Informationで提供されています。・・・PDFで。。。
とりあえずpdfminer.sixを使えばテキストを抽出することができるそうです*3 。pdfminer.sixのドキュメントはこちらで、GitHubはこちらです。
pip install pdfminer.six
一緒にダウンロードされるpdf2txt.pyを使えば簡単らしいです。
私はプログラミングが苦手なので、以降Jupyter notebookで実行しています。
まずPython3でpythonファイルを実行するため、エクスクラメーションマーク(!)をつけて!python3としています。pdf2txt.pyはAnacondaのbinの中にインストールされているようなので相対Pathを使ってpythonファイルを指定しました。
入出力は以下の通りです。
- 入力ファイル : ACSからダウンロードしたSupporting Information (「ml0c00556_si_001.pdf」)
- 出力ファイル(
-o):「SI.txt」
引数--page-numbersを使うと、抜き出したいページを指定できます(ページ番号をスペースを挟んで並べる)が、今回は目的のページ数が多く面倒なのですべて処理します。
!python3 ../../.pyenv/versions/anaconda3-5.3.1/bin/pdf2txt.py -o SI.txt ml0c00556_si_001.pdf
無事「SI.txt」ファイルができました。該当箇所はこんな感じ。
ただし、 ページ番号も同時に抽出されるので、座標の合間に改行やベージ番号が挟まっているためそのままでは使えません。 とりあえずコンフォマー1つ分座標があれば良いので手作業で取り除きました。
このままでもRgyrを計算することはできそうですが、折角ですので化学構造の取り扱いに適したファイル形式への変換を試みたいと思います。
座標のXYZ形式とPyMolでの確認
今回の座標の記載方法はXYZ形式に相当すると思います。化学の新しいカタチさんの記事「XYZ形式とZ-マトリックスは分子の立体構造を表す入力フォーマット」に詳しいのでご参照下さい。
上記記事によると、
という記載になっていれば良いようです。
論文の記載より今回の化合物1の組成式はC50H58N9O10F3S となっていますので原子の数は全部で131となります。 この情報を加えて「conformer_1.xyz」というファイルを作成しました。

xyzファイルが正しくできているか? 確認のためPyMolで描画してみます。
論文本文中のFig. 3とおおよそ同じ向きになるようにして並べてみました。
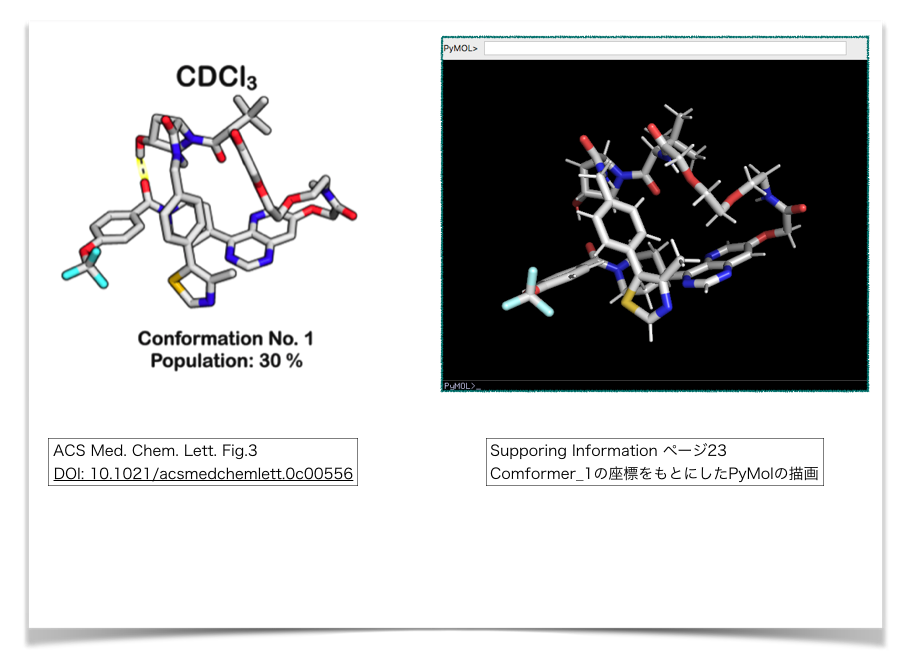
どうやらうまく情報を抽出できていそうです。
xyz形式は座標の情報しか入っていないようなので、結合次数の情報が無く、すべて単結合で表されているようです。・・・なるほど。
Open Babelによるファイル形式の変換と修正
欲しい情報を得られていそうなことがわかりました。ところで私はRDKitが好きなのですが、RDKitではxyzファイルを直接読み込むことができません。
そこで、Open Babelを用いてmolファイル形式に変換してみようと思います。ここではOpen Babelを扱えるPythonライブラリpybelを利用します。Pybelのドキュメントはこちらです。使い方は化学の新しいカタチさんの記事 「Open Babelを使って化学情報のファイル形式を変換」がとても参考になります。
扱えるファイル形式とその拡張子はドキュメントのこちら(Supported File Formats and Options)で確認できます。Jupyter notebook上でprint(pybel.informats)あるいはprint(pybel.outformats)として確認することもできます(入力 or 出力)。
早速、xyzファイルを読み込んでmolファイルに変換してみましょう!
readfile()でファイルを読み込めます。ただしこちらはジェネレーターオブジェクトです。今回は分子一つだけなので、__next__()で中身にアクセスして取り出しています。複数分子の時はforループで取り出せば良いらしいです。
import pybel single_obmol = pybel.readfile("xyz", "conformer_1.xyz").__next__()
出力はwrite()を使います。今回は1分子なのでそのままでOKです。
single_obmol.write('mol', './conformer_1.mol')
無事、conformer_1.molというファイルができました。
確認のためPyMolで描画してみましょう。先のxyzファイルの描画と並べてみます。
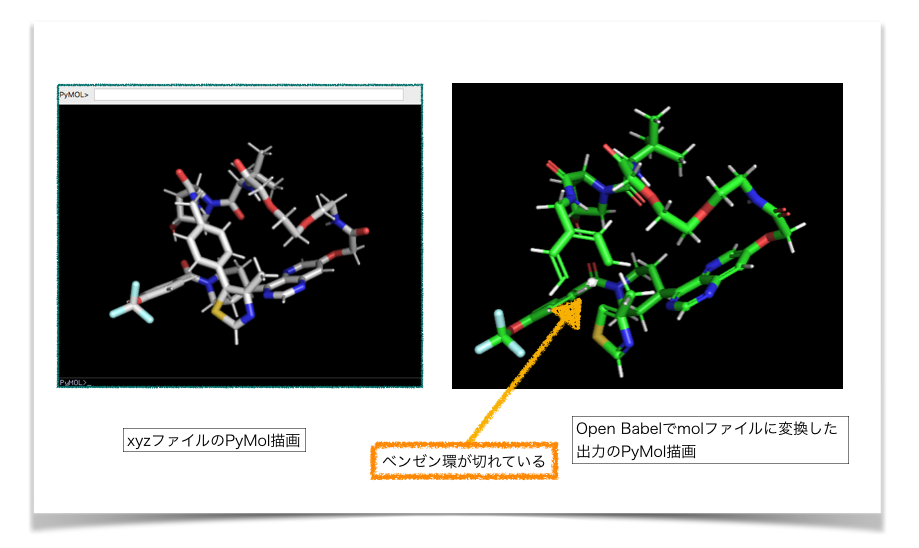
Open Babelで変換したMolファイルは単結合だけでなく2重結合も見えている一方で、ベンゼン環の結合が切れてしまっています。
どうしてこうなった??
おかしくなっている原子とその周りにAtom Identifiers: indexでラベルをつけてみました。*4。合わせてMolファイルの該当箇所を確認してみます。
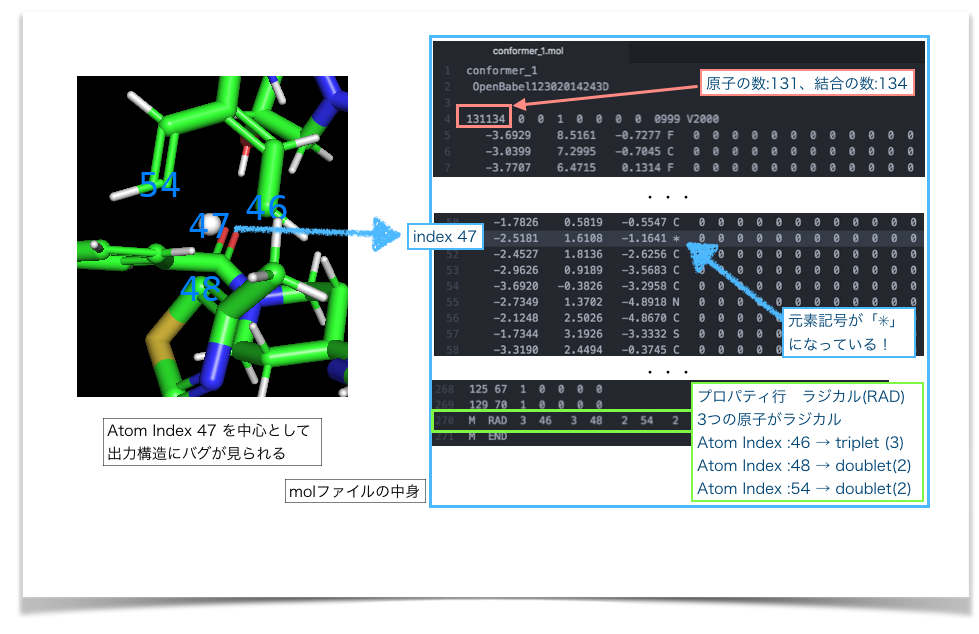
図のように、バグのある原子(Atom index: 47)は、molファイルのアトムリストで元素記号が「*」となっていました。また、結合リストでは47を中心とした結合関係が書かれていませんでした。結合次数が原子価に足りないAtom index 46、48、54はラジカルとして扱われてしまっています。
以上を踏まえて次のように修正してみました(conformer_1mod.mol)。
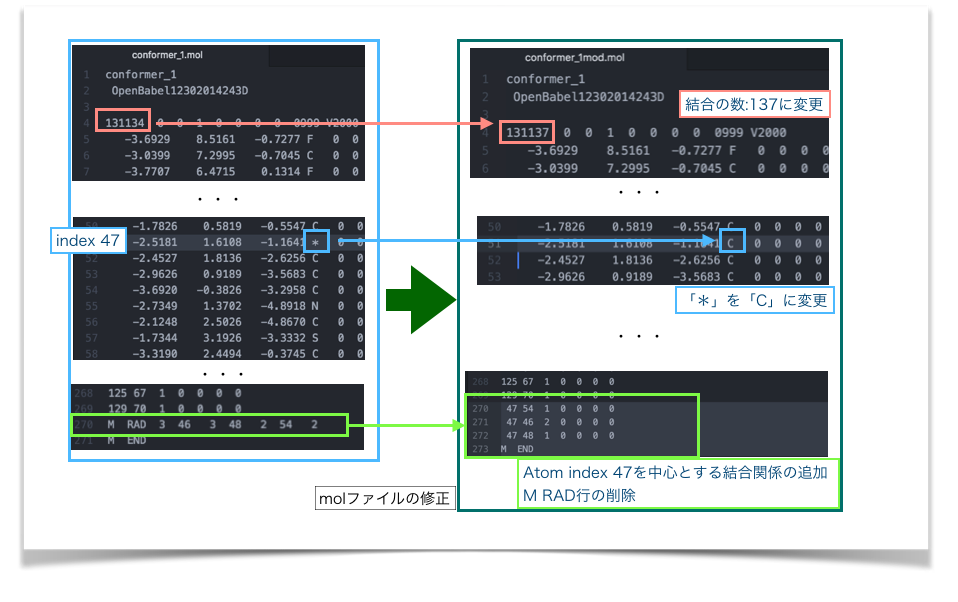
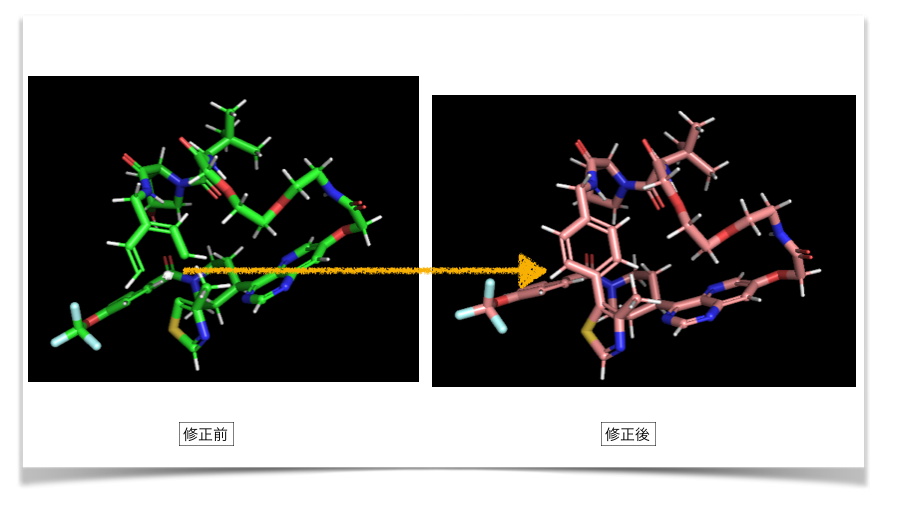
修正したmolファイルを再度PyMolで読み込み、修正前と比較してみました。
下図の通り、構造のバグが修正できているようです!!
修正したmolファイルがRDKitでも読み込めるか試してみましょう。
Molオブジェクトに変換したのち、py3DMolで3次元描画を行います。使い方は化学の新しいカタチさんの記事「py3Dmolを使って化学構造をJupyter上で美しく表示する」に詳しいです。
from rdkit import Chem from rdkit.Chem import AllChem, Draw from rdkit.Chem.Draw import IPythonConsole import py3Dmol conf1 = Chem.MolFromMolFile('./conformer_1mod.mol', removeHs=False) # 水素原子も必要 IPythonConsole.drawMol3D(conf1)
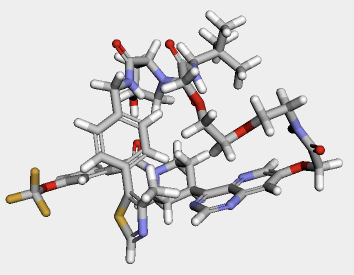
修正したmolファイルは無事RDKitで読み込むことができ、正しい構造を構築できていそうです。
ダメ押しで組成式も確認してみます。
from rdkit.Chem.rdMolDescriptors import CalcMolFormula formula = CalcMolFormula(conf1) print(formula) # C50H58F3N9O10S
Supporting Informationに記載のデータと同じ組成式が得られました。良かった良かった!!
なお、RDKitでxyzファイルを読み込めるようにするためのプログラムもあるようです。
- xyz2mol ( GitHub → https://github.com/jensengroup/xyz2mol)
- 使用例 Kaggle notebook → Using RDKit for Atomic Feature and Visualization
RDKitによるRadius of gyrationの計算
かなり遠回りしてしまいましたが、ここまでで論文からコンフォマーの座標の抽出と一般的な化学構造ファイルへの変換が終わりました。
本題、Radius of gyrationの計算を行ってみましょう!
PyMolWikiのRadius of gyrationを参考に、RDKitのMolオブジェクトから直接求められる形に修正を試みたいと思います。
まずは重心の座標(r_CM)を求めます。以下の手順でいいはず。。。
- Molオブジェクトから各原子の座標の取り出す
- 各原子の座標と原子量を掛け算して総和をとる
- 分子量で割り算
座標の取得は化学の新しいカタチさんの記事「RDKitによるコンフォマーの生成」を参考にさせていただきました。
こんな感じの関数を作ってみました。
def calc_r_CM(mol): from rdkit import Chem from rdkit.Chem import Descriptors import numpy as np # 各原子の原子量と座標を準備 mass_list = [] coordinate_list = [] mol_conf = mol.GetConformer(0) for i, (x, y, z) in enumerate(mol_conf.GetPositions()): # 原子量 atom = mol.GetAtomWithIdx(i) mass_list.append(atom.GetMass()) # 座標 coordinate_list.append((x,y,z)) # (x,y,z)それぞれに原子量x座標を計算し総和をとる r_CM_fraction = np.zeros(3) for m, (x, y, z) in zip(mass_list, coordinate_list): r_CM_fraction[0] += m*x r_CM_fraction[1] += m*y r_CM_fraction[2] += m*z # 分子量 MW = Descriptors.MolWt(mol) # 重心の座標 r_CM = r_CM_fraction / MW return r_CM
コンフォマー1のMolオブジェクトを投げ込んで重心の座標を求めてみます。
print(calc_r_CM(conf1)) # [-0.07220759 0.07596426 -0.02222654]
求まりました。ほぼ原点ですね。
続いて目的のRadius of gyration(RG)を求めます。先の関数と重複が多くなってしまいましたがとりあえずこんな感じ?
def calc_RG(mol): from rdkit import Chem from rdkit.Chem import Descriptors import numpy as np # 各原子の原子量と座標を準備 mass_list = [] coordinate_list = [] mol_conf = mol.GetConformer(0) for i, (x, y, z) in enumerate(mol_conf.GetPositions()): # 原子量 atom = mol.GetAtomWithIdx(i) mass_list.append(atom.GetMass()) # 座標 coordinate_list.append((x,y,z)) # 重心の座標 r_CM = calc_r_CM(mol) # 分母は分子量 MW = Descriptors.MolWt(mol) # 分子は(原子量 x (原子座標 - 重心の座標)^2 )の総和 r2_RG_fraction = 0 for m, (x, y, z) in zip(mass_list, coordinate_list): r2_RG_fraction += m * ((x - r_CM[0])**2 + (y - r_CM[1])**2 + (z - r_CM[2])**2) # 回転半径の2乗 r2_RG = r2_RG_fraction / MW # 回転半径 r_RG = np.sqrt(r2_RG) return r_RG
作成した関数でRadius of gyrationを求めてみます。
print(calc_RG(conf1)) # 5.481507768388569
約5.5Åと求まりました。さて正しい計算結果となっているでしょうか??
論文のFig.5 を引用します。
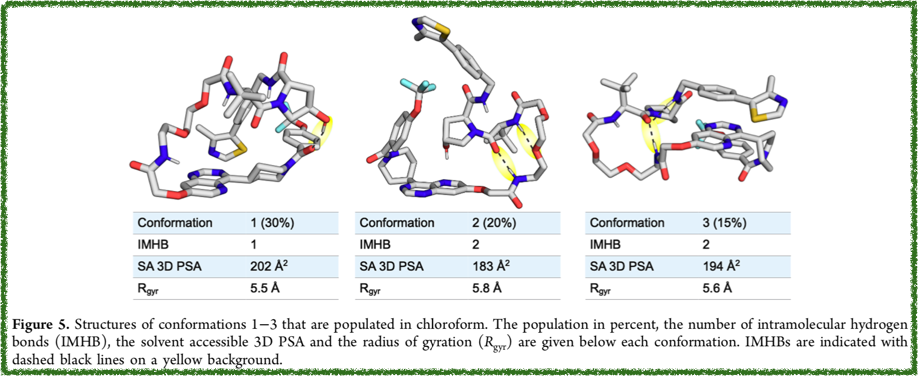
Conformation 1のRgyrは 5.5Åとなっています。どうやら合っていそうです。良かった!!
PyMolでradius of gyrationを描画
正しそうな結果は出ましたが、実際この数値の意味するものがうまくつかめません。
せっかく座標があるのでPyMolでコンフォマーと一緒に描画してみましょう!
Psudoatomを使えばいけそうです。
先に求めた重心の座標([-0.072, 0.076, -0.022] )とRgyr(5.48)を使って、PyMolのコマンドラインに以下を打ち込みました。
pseudoatom Rgyr, vdw=5.48, pos=[-0.072, 0.076, -0.022]
このままでは座標の点だけなのでShowでas meshとしてみました。

おおっ!!!
コンフォマーと重なるように球体が描かれました!!!!
確かにこれなら3次元的な構造を表す指標と言われても納得です!!!すげー!!!
まとめ
以上、今回は立体的な構造の指標、Radius of gyrationについて調べてみました。知らない指標だったのでPyMolWikiにも載っていてびっくりしました。
生物界隈でもよく使われる指標なのでしょうか??詳しい方教えていただけると幸いです。・・・あとNAFMISとSA 3D PSAについても教えてください。。。
また、実際に求める過程でxyz形式とその変換も試しました。予想外にOpen Babelで行った変換結果にはバグがありました。 ファイル形式の変換によく使われるソフトと聞いていたので、あれっ!!!ちゃうやん!!!って感じでした。
xyz形式は座標と原子の種類の情報しかないため、情報量が少ないことがバグが生じた原因なのかもしれません。
情報なんてなんぼ合ってもいいですからね!
いずれにせよファイル形式の変換は注意しないといけないということがわかって良かったです。
今回もいろいろと間違いが多そうなのでご指摘いただけると幸いです。
ではでは!
*1:どういうことかさっぱりわかりません。 ref. J. Am. Chem. Soc. 1995(117)1027
*2: Supportgint Informationより、ソフトウェアはシュレーディンガー社のMacromodelのようです
*3:【Python】pdfファイルからテキストを超簡単に抽出する方法
*4:PyMolのAtomラベルのサイズと色はPyMol Wiki:Labelを参考に変えています
ChEMBLのHELMモノマーライブラリーを解析した話 〜XMLをDataFrameに変換して部分構造検索〜
マクロ分子の表現HELMについて調べています。HELMは階層的な構造をとっており、モノマー (ex. アミノ酸)を組み合わせてポリマー (ex.ペプチド)を表現します。
HELMの特徴はその表現の拡張性にあり、モノマーライブラリーにオリジナルなモノマーを追加することで非天然な構造を表現することもできます。
一方で、HELM表現は略記(ID)を用いるので、モノマーライブラリーが共有されていない場合、同一のIDで異なるモノマーを指定する危険性があるため、モノマーライブラリを把握しておくことが重要となりそうです。
そこで今回はHELMも扱っているおなじみのデータベース、ChEMBLでどのようなモノマー情報が格納されているか、探ってみたいと思います。
具体的にはXMLファイルで提供されているモノマーライブラリーを読み込んでPandasDataFrameに変換することを目指します。
つまりXML良くわからない私の単なるお勉強メモ!!
- ChEMBL HELM モノマーライブラリーの取得
- XMLの中身
- XML宣言と名前空間
- ElementTreeでXMLを解析
- モノマー情報の取得
- DataFrameへの変換
- DataFrame内の構造式検索
- まとめ
ChEMBL HELM モノマーライブラリーの取得
ChEMBL のHELM モノマーライブラリーをはHELM表記のある化合物のCompound Report Cardから取得することができます。
例えば、GnRH(性腺刺激ホルモン放出ホルモン)アンタゴニストであるアバレリクス(Abarelix)の場合、Compound Report CardにおけるHELM表記は以下のようになっています。
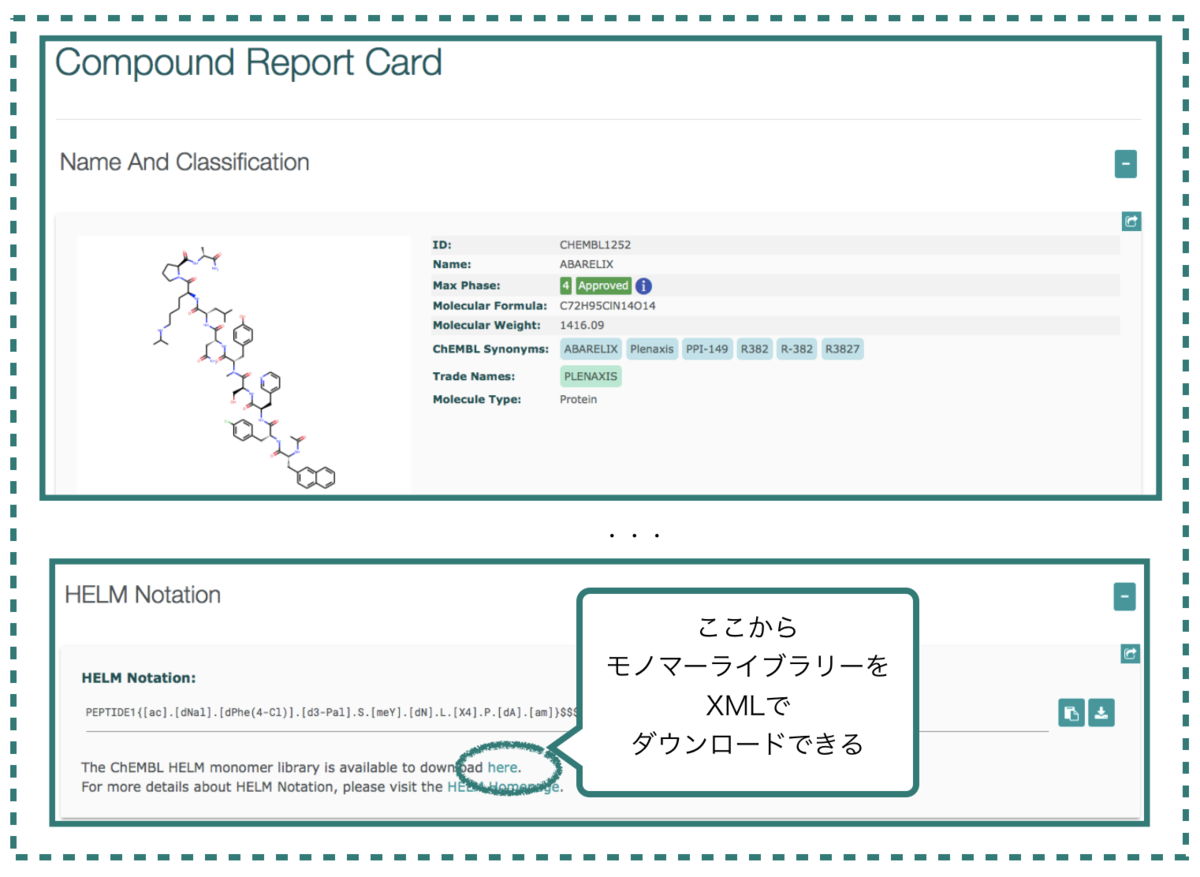
図の真ん中あたりの「here」 というところをクリックするとxmlファイルのダウンロードが始まります。
2020/11/23現在「chembl_27_monomer_library.xml」というファイルが提供されています。
XMLの中身
XMLについてよくわかっていないので、順番にファイルの中身を見ていきたいと思います。
話がそれますがこちらの記事(Macs in Chemistry 「Determining the Amino Acids in a collection of peptides」)でChEMBLモノマーライブラリーXMLの解析事例が紹介されているので、正しい情報が欲しい方はご参照ください。
閑話休題。
とりあえずテキストエディタで開くと中身はこんな感じの構造になっています。

HELMの記載方法同様、モノマーライブラリも階層的な構造となっているようです。
- <PolymerList>
- <Polymer>
- <monomer>
という順にタグが入れ子構造(ネスト)になっており、<monomer> ~ </monomer>ごとに各モノマーの情報が入っているようです。
monomerに記載されている情報は、Pistoia Alliance HPのHELMの解説におけるMonomerと見比べてみると分かりやすいですね。

上図のテーブルに該当するタグがXMLに含まれているようです。
それでは具体的にどのようなモノマーが含まれているのか、その総数や各モノマーの特徴解析を進めていきたいと思います。
・・・・が、早速冒頭2行から意味がわかりませんでした。
XML宣言と名前空間
ファイルの冒頭2行、以下のように記載されています。
<?xml version="1.0" encoding="UTF-8"?> <MonomerDB xmlns="lmr" xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance">
1行目はXML宣言というもので、使用するXMLのバージョンや文字コードについて記述してあるそうです。*1
この場合バージョン1.0、文字コードUTF-8となっているようです。
2行目、タグの中にある「xmlns="lmr"」という箇所は、XMLの名前空間を表しているようです。*2
XMLマスターというこちらのページの説明が分かりやすかったです。
XML名前空間とは、XML文書内にある「定義内容は異なるが同じ名前の要素名または属性名」を区別し、名前の衝突を回避するための仕組みです。(XMLマスターポイントレッスン 〜ベーシック編〜 第11回 XML名前空間 より引用)
ここでは接頭辞(prefix)を使わずに名前空間宣言が行われているため、デフォルトの名前空間に相当する記載となっています。
従って、MonomerDBという要素に対して、lmrをデフォルトの名前空間識別子(URI: Uniform Resource Indentifier)として宣言していることになり、
基本的には全てのタグが{lmr}タグという形に展開されることになります。
一方で、2行目後半「xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance"」 という箇所はXMLのスキーマ定義に関わっているようです。xsiという接頭辞にXML Schema instanceをURIとして宣言しています。*3
どういうこっちゃ?
ElementTreeでXMLを解析
「入門 Python3」によると、XMLの解析にはとりあえずElementTreeを使えば良いとのことなので、試してみます。
ライブラリをインポートした後、まずはparseでファイルを読み込みます。その後getroot()でルートとなるノードを取得して処理を行えば良いそうです。*4
import xml.etree.ElementTree as ET tree = ET.parse('./chembl_27_monomer_library.xml') root = tree.getroot()
タグはtag、属性はattribに入っている、とのことなので確認してみます。
print(root.tag) # {lmr}MonomerDB print(root.attrib) # {}
先に見た通り、デフォルトの名前空間がlmrとなっているため、タグのURIとして{lmr}が付与されています。
また、MonomerDBタグは名前空間の宣言以外の属性がないのでattribは空となっているようです。
子ノードを取得してみます。*5イテレート可能、とのことなのでforループでタグを表示させてみます。
for child in root: print('タグ: ', child.tag, '属性: ', child.attrib) # タグ: {lmr}PolymerList 属性: {} # タグ: {lmr}AttachmentList 属性: {}
PolymerListの他にAttachmentListというタグがあるようです。
PolmerLystのもう一つ下の階層にアクセスします。インデックスを利用してみます。
polymerlist = root[0] print(len(polymerlist)) for child in polymerlist: print(child.tag, child.attrib) # 1 # {lmr}Polymer {'polymerType': 'PEPTIDE'}
PolymerTypeはPEPTIDEのみでした。HELMには他に RNA、SAC、CHEMといったPolymerTypeがありますが、ChEMBLではPEPTIDEのみしか含まれていないようです。
モノマーライブラリーがペプチドのみ、ということがわかったので含まれるモノマーは全てアミノ酸(誘導体)となっていると思われます。
いくつのモノマーが含まれているでしょうか?名前空間のURIに気をつけて{lmr}Monomerの全ての要素をfindall()で取得します。
polymer = root[0][0] monomer_elements = polymer.findall('{lmr}Monomer') print(len(monomer_elements)) # 2851
全部で2851個のモノマーが登録されているようです。PEPTIDEのモノマーってそんなに種類あるんですか???
上記はindexで階層を辿っていますがXPath機能を使うこともできるそうです。.//を使うことで木全体から要素を選択することができます。*6
monomer_elements_2 = root.findall(".//{lmr}Monomer") print(monomer_elements == monomer_elements_2) # True
先に取り出したものと一致することが確認できました。
モノマー情報の取得
ElementとしてMonomerを取り出すことができました。その中身を表示して見ます。
タグで囲まれた内容は.textで確認することができます。
mon = monomer_elements[0] monomer_tag_list = [] for m in mon: monomer_tag_list.append(m.tag) print(m.tag, m.text) # {lmr}MonomerID X2484 # {lmr}MonomerSmiles CC(=O)SCCCC[C@H](N[*])C([*])=O |$;;;;;;;;;;_R1;;_R2;$| # {lmr}MonomerMolFile H4sIAAAAAAAAAI2TTU7DMBCF9z7FSHTLaMb/swbUFQW1iAtUCPX+F2BsN7ZTUYgVRe/Fn1/G48QAnM6Xj6/vy5mYLQcmS/bZGGAP7ACoXlzvY4gIfFoiMsUxOpVNSWiKkMqzJ2gRNxxh+oMjtJFiUxzITdzbzFmMtEoZ6jRzDmXTewPae3krLmLYlJcwlUb9mydInbuTp5tdxFgTc+uCDZSnNYc5OyOFhcte/MQdHwbHhG5LrYN75NturfIYvZPryQWR9PsZct0XN6PKtX024/uMqtCNqtiNqtSNqtyNLCZWw7TMqGKeMO4VqCqf/DD+Ws4rwHH/3mplqEQBwZaZl4P+Ljsd5gd2o4VdTQMAAA== # {lmr}MonomerType Backbone # {lmr}PolymerType PEPTIDE # {lmr}NaturalAnalog X # {lmr}MonomerName X2484 # {lmr}Attachments
モノマーを表す特徴として8つの情報が含まれています。先に図で示したXMLファイルの中身と同様の内容がtextで取り出されているようです。
最後、Attachmentは更に深い階層に情報が書かれています。一つ一つ階層を辿るのが面倒なので手っ取り早くiter()での処理を試してみます。
Elementオブジェクトはiter()で現在の要素を根とする木のイテレータが作成できるそうです。*7
Att = mon.find('{lmr}Attachments') for Att_el in Att.iter(): print(Att_el.tag, Att_el.text) # {lmr}Attachments # {lmr}Attachment # {lmr}AttachmentID R1-H # {lmr}AttachmentLabel R1 # {lmr}CapGroupName H # {lmr}CapGroupSmiles [*][H] |$_R1;$| # {lmr}Attachment # {lmr}AttachmentID R2-OH # {lmr}AttachmentLabel R2 # {lmr}CapGroupName OH # {lmr}CapGroupSmiles O[*] |$;_R2$|
2箇所のAttachmentについてそれぞれの下の階層の情報まで取得することができました。
IDやLabel、CapGroupの情報が含まれているようです。ここでもSMILESはCXSMILESのようですね。
DataFrameへの変換
Monomer情報の取得ができるようになったので、より扱いやすくするためモノマーライブラリーをDataFrameに変換してみたいと思います。
具体的にはRDKitのPandasTools*8を利用して、各行に各Monomerの情報とMolオブジェクトを含むDataFrameを作成します。作成したDataFrameは部分構造検索などが利用できるので、簡易的なデータベースとして使えるはず!
まず、8つの特徴のうち、MonomerMolFileとAttachmentsを除いた以下の6つを対象として、DataFrameのカラムとします。
- MonomerID
- MonomerSmiles
- MonomerType
- PolymerType
- NaturalAnalog
- MonomerName
monomer_tag_list.remove('{lmr}MonomerMolFile') monomer_tag_list.remove('{lmr}Attachments') # 見やすさのため{lmr}を取り除いて列名とする column_names = [s.replace('{lmr}', '') for s in monomer_tag_list]
必要なライブラリを読み込みます。
from rdkit import rdBase, Chem from rdkit.Chem import AllChem, Draw, rdDepictor, PandasTools from rdkit.Chem.Draw import IPythonConsole import pandas as pd # DataFrameの準備 df = pd.DataFrame(columns = column_names )
モノマーごとに取得した情報をリストに格納後、pd.Seriesに変換して、DataFrameに追加していきます。
for m in monomer_elements: data = [] for tag in monomer_tag_list: e = m.find(tag) data.append(e.text) s = pd.Series(data, index=column_names) df = df.append(s, ignore_index=True) print(df.shape) # (2851, 6)
2851個のモノマーを含むDataFrameが作成できました!
中身を確認してみます。
df.head()
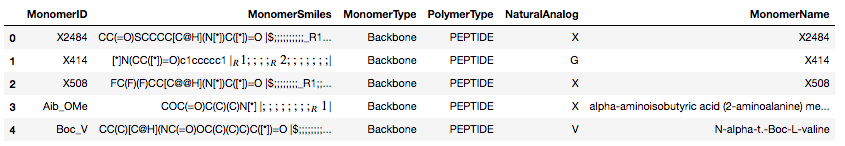
きちんと情報が入っていそうです。
RDKitのPandasToolsを使って各行のMonomerSMILESからROMolオブジェクトを作成します。
PandasTools.AddMoleculeColumnToFrame(df, 'MonomerSmiles')
df.head()
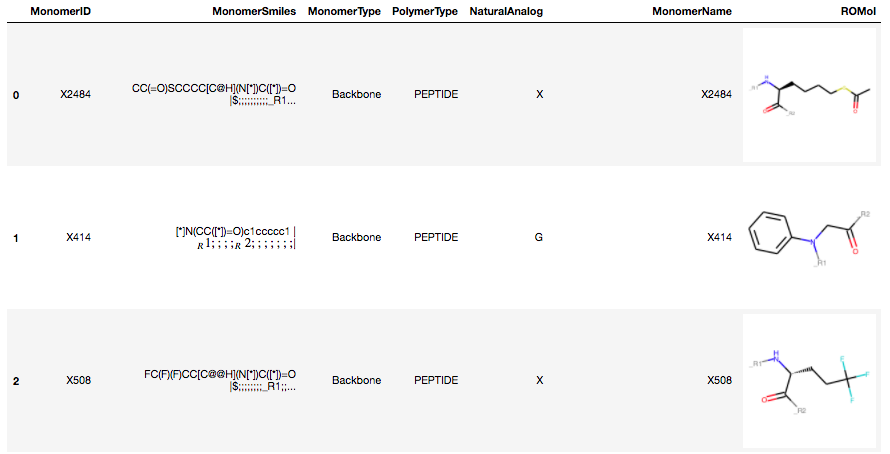
構造式データを含むDataFrameが無事作成できました!
DataFrame内の構造式検索
ではPandasToolsで作成したDataFrameをが構造式データベースとして利用可能か? 部分構造検索等、試していきましょう!
例としてチロシンの誘導体にどのような構造が含まれているか検索してみます。
まずは簡単にNaturalAnalogとしてYが設定されているものを抜き出してみます。
Y_derivatives = df.query('NaturalAnalog == "Y"') print(Y_derivatives.shape) # (176, 7)
176個のチロシン関連構造が含まれているようです。L-チロシンそのものの情報を確認してみます。
Y_derivatives.query('MonomerName == "Tyrosine"')
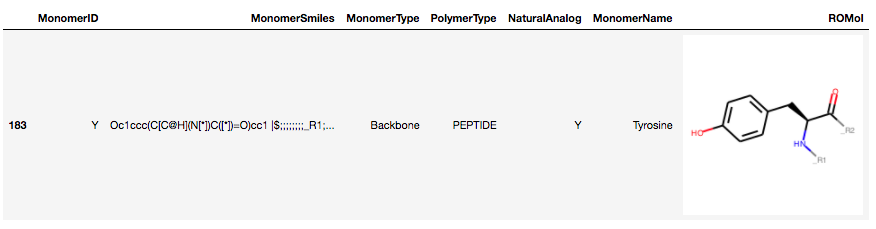
L-Tyrosineがどのように格納されているかがわかったので、この構造情報をつかって部分構造検索を行ってみます。
元のDataFrameから「>= (ge比較演算子)」を使って検索します。
# L-tyrosineのROMolオブジェクトをDataFrameから取得 L_ty = df.iat[183, 6] # ge比較演算子で部分構造マッチ L_ty_sub = df[df.ROMol >= L_ty] print(L_ty_sub.shape) # (77, 7) L_ty_sub.head()
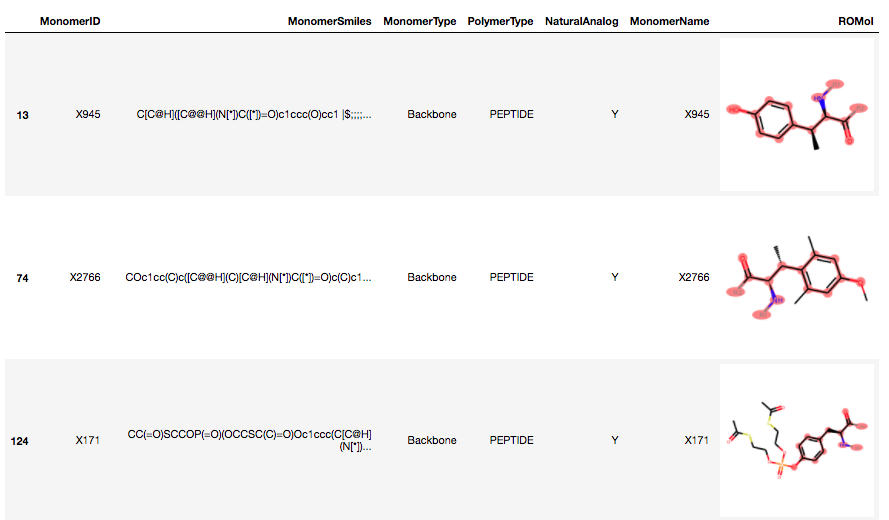
L-tyrosineを部分構造として含むものは77個見つかりました。先にNaturalAnalog == Yとしたものは176個でしたので、チロシンのアナログのなかには元の部分構造自体にも変化が加わっているものがあるようです。
では逆にL-tyrosineと部分構造が一致したもののNaturalAnalogは全てYとなっているのでしょうか?
Analog_vc = L_ty_sub['NaturalAnalog'].value_counts() Analog_vc # Y 72 # X 5 # Name: NaturalAnalog, dtype: int64
NaturalAnalogがXとなっているものも5つあるようです。
L_ty_sub_X = L_ty_sub.query('NaturalAnalog == "X"')
L_ty_sub_X.head()

NaturalAnalogがXとなっていたものは、L-tyrosineと一致する構造をもちつつ、ビシクロ環構造となっているもののようです。
こんな構造もモノマーに入ってるんですね。予想以上に非天然です。。。
試しに一番下のモノマー「MonomerID X316」を含む構造にどのようなものがあるかChEMBLで検索してみました。
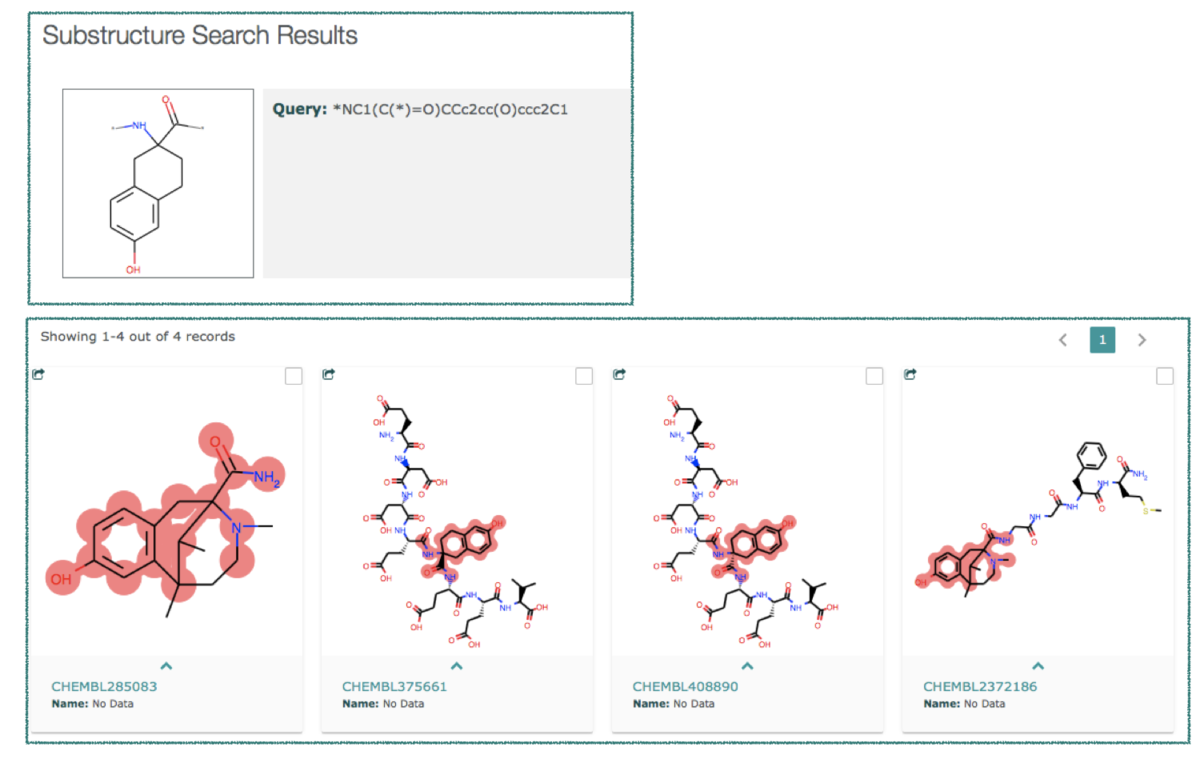
左から2番目、化合物「CHEMBL375661」の情報元の文献は以下でした。
Spatial Conformation and Topography of the Tyrosine Aromatic Ring in Substrate Recognition by Protein Tyrosine Kinases
J. Med. Chem. 2006, 49, 6, 1916–1924
この部分構造が含まれているのも納得なタイトルですね!
まとめ
以上、今回はChEMBLデータベースが提供しているHELMのモノマーライブラリーの中身を解析してみました。
アミノ酸の誘導体だけで3000個近くものモノマーがあるというのはびっくりです。
また、XMLの解析の練習をかねてDataFrameへの変換を行ってみました。RDKitのPandasToolsを使うことで部分構造検索もできるので、うまく使うことができれば簡単なデータベース感覚で利用できそうです。比較演算子が使えることがここまで便利とは思いませんでした。もうちょっと良い使い方があるか考えてみよう。
今回も色々と間違いが多そうです。ご指摘いただければ幸いです。
*1:Let'sプログラミング 「XML宣言を記述する」
*2:ELementTree XML API 「名前空間のあるXMLの解析」
*3:都元ダイスケ IT-PRESS 「XMLのスキーマの定義の仕方(2)」
*4:参考 LIFE WITH PYTHON 「ライブラリ:ElementTree」
*6:ELementTree XML API 「XPath サポート 」
*7:ELementTree XML API 「Element オブジェクト 」
*8:化学の新しいカタチ 「RDKitのPandasToolsでデータ分析を加速する 」
CXSMILESでSMILESに情報を追加する話
マクロ分子の表現HELMについて調べているうちに見慣れないSMILES表現に出会いました。Pistoia Alliance HPのHELMの解説でMonomer (Alanine) の表現として記載されていたものです。

色々と調べた結果おそらくCXSMILESなのかな?という結論に達しました。
というわけで、CXSMILESについての記事です。ついでにフェロセンをどう表すか問題についても遊んでみました。
CXSMILES
CXSMILES(ChemAxon Extended SMILES)はChemAxon社によるSMILESの拡張表現です(そのまんま)。
簡単にいうとSMILES文字列のあとに分子の特徴を追加で記載する表現形式です。基本的な形式は以下の通り
SMILES_String |<feature1>,<feature2>,...|
SMILESではスペースやタブで区切って書かれた情報をコメントとして無視することになっているそうです。これを利用して、区切りの後にバーティカルバー「|」で挟む形で特徴を記載し、+αの情報を追加する、というのが基本的な内容です。
記載できる特徴としては原子の座標、アトムラベル、配位結合・水素結合、立体配置といったものがあります。
ChemAxson社のこちらのページ (ChemAxson Extended SMILES and SMARTS - CXSMILES and CXSMARTS )に追加できる特徴やその記載方法が書かれているのでご参照ください。*1
RDKitとCXSMILES
CXSMILESはRDKitでも取り扱うことができます。The RDKit BookのCXSMILES extentions項の記載をChemAxonの説明と比較しつつ表にしてみました。*2
| 構文解析(parse)できる特徴 | 記載方法 | MolToCXSMILESで書き出せるか? |
|---|---|---|
| 原子座標 atomic coordinate |
(x,y,z);(x,y,z); |
できる |
| 原子価 atomic values |
$_AV: |
できる |
| 原子のラベル/エイリアス | $で挟み、各ラベルは;で区切る |
できる |
| 原子のプロパティ | atomprop:0.key1.value1:0.key2.value2:1.key3.value3アトムインデックス、キー、バリューを1セットにコロンで区切る |
できる |
| 配位結合 | C:結合の最初の原子のインデックス.結合のインデックス, RDKitでは2重結合に翻訳される |
|
| ラジカル | 1価のラジカル中心は^1:の後、2価のラジカル中心は ^1:の後... |
できる |
| 立体化学表現の拡張 enhanced stereo |
絶対立体配置 a:<atom index>,... OR o:<atom index>,...AND &:<atom index>,... |
できる |
| Link node | LN:<atom index>:<minimum repetition>.<maximum repetition>.<first outer atom index>,<second outer atom index>,... |
|
| 多重中心 multi-center attachments |
m:<multicenter atom index>:<atom index>.<atom index>... |
|
| 環結合数の指定 rinb bond count specification |
rb |
|
| 非水素置換数の指定 | s |
|
| 不飽和の指定 | u |
RDKitで原子のラベル/エイリアスとして認識されるものは_AP、star_e、Q_e、QH_p、AH_P、X_p、XH_p、M_p、MH_p、*です。
enhanced stereoはRDKitではStereoGroupsに変換されます。ABS、AND、ORの説明と利用方法はThe RDKit BookのSuppoer for Enhanced Stereochemistryの項に記載があります。
Link Nodeは実例があまりなく何を表すのかわかりませんでした。ご存知の方教えていただけると嬉しいです。
Pistoia Alliance アラニンモノマーの解釈
CXSMILESの記法だけを並べてもよくわからないので、実例として冒頭のアラニンを見直してみます。

まず、SMILES文字列中のアスタリスク(*)は「水素原子以外の全ての原子」を表すワイルドカードなのでSMARTSにあたると思います。*3
スペースを挟んで追記されている特徴は以下の通りです。
- 「r」 相対的立体配置(relatice stereoconfiguration)を表します。
- 「$;;;_R1;;_R2;$」 原子のラベル/エイリアスを表します。
原子一つずつをセミコロンで区切ってあらわすので、SMILESにおける4番目と6番目の(水素以外の)原子、つまり2つのワイルドカード(*)に対して「_R1、_R2」というラベルを与えています。
このCXSMILES表記はHELMモノマーのアラニンを表すので、「ペプチド結合の相手に何が来ても良いようにSMILESを表現する」という要件を満たすように拡張表記が活用されている、という様子ですね。
Atom label/aliaseについてはRDKitでも取り扱えるとのことですので、早速読み込んでみましょう。
from rdkit import rdBase, Chem from rdkit.Chem import Draw from rdkit.Chem.Draw import IPythonConsole print('rdkit version: ', rdBase.rdkitVersion) # rdkit version: 2020.03.2 alanine_monomer = Chem.MolFromSmiles('C[C@H](N[*])C([*])=O |r,$;;;_R1;;_R2;$|') alanine_monomer

無事、_R1や_R2といったラベルを含めたMolオブジェクトを生成することができました。原子の情報を確認してみます。*4
# 原子の取得 atoms = [a for a in alanine_monomer.GetAtoms()] print('atom index 3: Symbol: ', atoms[3].GetSymbol()) print('atom index 3: Properties: ', atoms[3].GetPropsAsDict()) # atom index 3: Symbol: * # atom index 3: Properties: {'dummyLabel': '*', 'atomLabel': '_R1', '__computedProps': <rdkit.rdBase._vectNSt3__112basic_stringIcNS_11char_traitsIcEENS_9allocatorIcEEEE object at 0x11ea3ae10>, '_CIPRank': 1}
ワイルドカードにはdummy labelというKeyが、_R1にはatomLabelというKeyが割り当てられているようです。
セミコンマによる区切りがわかりづらいので数字をそのまま割り当ててみます。
alanine_fig = Chem.MolFromSmiles('C[C@H](N[*])C([*])=O |$0;1;2;3;4;5;6$|')
alanine_fig

なるほど。
出力と読み込み
CXSMILESの動作が分かってきたので、もう一例CXSMILESの読み書きを行ってみます。
例としてRDKit Blog「 New drawing options in the 2020.03 release」で取り上げられているジクロフェナクをお借りします。
from rdkit.Chem.Draw import rdMolDraw2D from rdkit.Chem import rdDepictor rdDepictor.SetPreferCoordGen(True) from rdkit.Chem.Draw import IPythonConsole from IPython.display import SVG diclofenac = Chem.MolFromSmiles('O=C(O)Cc1ccccc1Nc1c(Cl)cccc1Cl') cp = Chem.Mol(diclofenac) cp.GetAtomWithIdx(2).SetProp("atomNote","pKa=4.0") d2d = rdMolDraw2D.MolDraw2DSVG(350,300) d2d.drawOptions().setHighlightColour((0.8,0.8,0.8)) d2d.DrawMolecule(cp,highlightAtoms=[2]) d2d.FinishDrawing() SVG(d2d.GetDrawingText())

カルボキシル基の酸素原子にpKaのプロパティをセットしています。
これをCXSmilesに変換してみます。
cp_cxsmi = Chem.MolToCXSmiles(cp) print(cp_cxsmi) # 'O=C(O)Cc1ccccc1Nc1c(Cl)cccc1Cl |atomProp:2.atomNote.pKa=4.0|'
SMILES文字列に続いてatomPropが加わっています。Atom Index 2の原子に対して、atomNoteというKeyにpKa=4.0というValueがセットされています。
このCXSMILESから再度Molオブジェクトを構築してみます。
cp_reconst = Chem.MolFromSmiles(cp_cxsmi) cp_reconst

再構築したMolオブジェクトでも原子のプロパティがうまく認識されているようです。
線形表記にプロパティを追加でき、再構築しても情報が再現できるというのは多数の分子を取り扱う際にデータ量の節約になりそうです。
フェロセン問題
つづいて話題をかえて「フェロセンどうやって表記するの?」問題に取り組みます。
唐突感がありますが、フェロセンはどうもSMILESでは表しづらい分子だそうです。
フェロセンのSMILES表記
SlideShareに公開してくださっていた資料 「第11回分子化学討論会 PubChemQCプロジェクト: 分子データベース構築と機械学習による電子構造の推定」のスライド p13には「SMILESで表現できない例」というタイトルでFerroceneが例示されています。
スライドには表現例として以下の2つが記載されています。2つめはPubChem(Ferrocene) の表記と同じです。
- C12C3C4C5[Fe]23451234C5C1C2C3C45
- [CH-]1C=CC=C1.[CH-]1C=CC=C1.[Fe+2]
それぞれRDKitでMolオブジェクトに起こしてみましょう。まずは1つ目。。。
Fer_1 = Chem.MolFromSmiles('C12C3C4C5C1[Fe]23451234C5C1C2C3C45')
Fer_1
 !
!
・・・・スターウォーズにこういう飛行機あった気がする。
for b in Fer_1.GetBonds(): print('{}: {}'.format(b.GetIdx(), b.GetBondType())) # 0: SINGLE # 1: SINGLE # 2: SINGLE # 3: SINGLE # 4: SINGLE # 5: SINGLE # 6: SINGLE # 7: SINGLE # 8: SINGLE # 9: SINGLE # 10: SINGLE # 11: SINGLE # 12: SINGLE # 13: SINGLE # 14: SINGLE # 15: SINGLE # 16: SINGLE # 17: SINGLE # 18: SINGLE # 19: SINGLE
こちらの表現ではFe(II)イオンとシクロペンタジエニルアニオンの結合が明示されている一方で、20本全ての結合が単結合(SINGLE)となっており、芳香属性を表すことができていません。
つづいて2つ目の表現です。
Fer_2 = Chem.MolFromSmiles('[CH-]1C=CC=C1.[CH-]1C=CC=C1.[Fe+2]') d2d = rdMolDraw2D.MolDraw2DSVG(200,200) d2d.drawOptions().addBondIndices=True d2d.DrawMolecule(Fer_2) d2d.FinishDrawing() SVG(d2d.GetDrawingText())

for b in Fer_2.GetBonds(): print('{}: {}'.format(b.GetIdx(), b.GetBondType())) # 0: AROMATIC # 1: AROMATIC # 2: AROMATIC # 3: AROMATIC # 4: AROMATIC # 5: AROMATIC # 6: AROMATIC # 7: AROMATIC # 8: AROMATIC # 9: AROMATIC
分かりやすさのためい結合のインデックスも表記しています。描画ではケクレ構造となっていますが、2つめの表現ではシクロペンタジエニルアニオンの結合がすべて芳香属性として認識されています。
一方で、こちらではFe(II)イオンとの間の結合が表現できていません。
SMILESの拡張表現を使えばうまくフェロセンを表現できるのか?検証してみましょう。
CXSMILESの配位結合表記
ChemAxonのCXSMILES解説には、「CXSMILESの一部として現れるSMILESが、必ずしも標準的なSMILESに一致しない」という事例としてフェロセンが挙げられています。
- plain SMILES: [Fe].c1cccc1.c1cccc1
- CXSMILES: c12c3c4c5c1[Fe]23451234c5c1c2c3c45 |C:4.5,0.6,1.7,2.8,3.9,7.12,6.10,9.16,10.18,8.14|
但しこれらはどちらもRDKitではエラーが出てしまい扱うことができません。RDKitではシクロペンタジエニルアニオン部位を芳香属性のc1cccc1であらわすとケクレ化ができないとなってしまいます。sanitize=Falseの設定をすることでオブジェクを作ることはできますがDrawでエラーがいっぱい出てきます。
cpa = Chem.MolFromSmiles('c1cccc1', sanitize=False) cpa # エラーメッセージは省略

話を簡単にするために、ここではCXSMILESの配位結合の表現の利用箇所だけを検証したいと思います。
配位結合の表現は「C:結合の最初の原子のインデックス:結合のインデックス」ですので、Feに結合するBondのインデックスと結合に関わる原子のインデックスを取得します。原子のインデックスは配位結合の出発点、FeではなくCとなるようにします。
Fe_connected_bonds = [] C_idx_list = [] for b in Fer_1.GetBonds(): bond_idx = b.GetIdx() begin_atom = b.GetBeginAtom() begin_symbol = begin_atom.GetSymbol() begin_idx = begin_atom.GetIdx() end_atom = b.GetEndAtom() end_symbol = end_atom.GetSymbol() end_idx = end_atom.GetIdx() if begin_symbol == 'Fe': Fe_connected_bonds.append(bond_idx) C_idx_list.append(end_idx) elif end_symbol == 'Fe': Fe_connected_bonds.append(bond_idx) C_idx_list.append(begin_idx) print(Fe_connected_bonds) # [4, 5, 11, 12, 13, 14, 15, 16, 17, 18] print(C_idx_list) # [4, 6, 7, 0, 8, 1, 9, 2, 10, 3]
追記すべき配位結合の表現は以下となります。
dative_bonds = 'C:' n = len(Fe_connected_bonds) for i in range(n-1): dative_bonds = dative_bonds + str(C_idx_list[i]) + '.' + str(Fe_connected_bonds[i]) + ',' dative_bonds = dative_bonds + str(C_idx_list[n-1]) + '.' + str(Fe_connected_bonds[n-1]) print(dative_bonds) # C:4.4,6.5,7.11,0.12,8.13,1.14,9.15,2.16,10.17,3.18
従ってCXSMILESはこんな感じになりそうです。
Fer_1_CXSMILES = 'C12C3C4C5C1[Fe]23451234C5C1C2C3C45 |C:4.4,6.5,7.11,0.12,8.13,1.14,9.15,2.16,10.17,3.18|'
Fer_1_CX = Chem.MolFromSmiles(Fer_1_CXSMILES)
Fer_1_CX

できました! あいかわらずシクロペンタジエニルアニオンは単結合のままですが配位結合はそれとして認識できているようです!
念のため確認...
for b in Fer_1_CX.GetBonds(): print('{}: {}'.format(b.GetIdx(), b.GetBondType())) # 0: SINGLE # 1: SINGLE # 2: SINGLE # 3: SINGLE # 4: DATIVE # 5: DATIVE # 6: SINGLE # 7: SINGLE # 8: SINGLE # 9: SINGLE # 10: SINGLE # 11: DATIVE # 12: DATIVE # 13: DATIVE # 14: DATIVE # 15: DATIVE # 16: DATIVE # 17: DATIVE # 18: DATIVE # 19: SINGLE
CXSMILESから起こした構造は配位結合をDative bondとして認識することができているようです。
RDKitで配位結合を形成する
次に、RDKitで配位結合を形成し、それをCXSMILESに変換するという方法を試してみます。
シクロペンタジエニルアニオンとFeの間に結合の無かったオブジェクトに対して配位結合を追加していきます。 まずは原子のインデックスを確認します。
d2d = rdMolDraw2D.MolDraw2DSVG(200,200) d2d.drawOptions().addAtomIndices=True d2d.DrawMolecule(Fer_2) d2d.FinishDrawing() SVG(d2d.GetDrawingText())

Fe (Atom Index 10)に対して、シクロペンタジエニルアニオン (Atom Index: 0 ~ 9)から配位結合を形成すれば良いようです。
結合を編集するために、RWMolにオブジェクトを変換します*5
Fer_2_RW = Chem.RWMol(Fer_2) type(Fer_2_RW) # rdkit.Chem.rdchem.RWMol for i in [0, 1, 2, 3, 4, 5, 6, 7, 8, 9]: Fer_2_RW.AddBond(i,10, Chem.BondType.DATIVE) Fer_2_RW

for b in Fer_2_RW.GetBonds(): print(b.GetIdx(), b.GetBeginAtomIdx(), b.GetEndAtomIdx(), b.GetBondType()) # 0 0 1 AROMATIC # 1 1 2 AROMATIC # 2 2 3 AROMATIC # 3 3 4 AROMATIC # 4 5 6 AROMATIC # 5 6 7 AROMATIC # 6 7 8 AROMATIC # 7 8 9 AROMATIC # 8 4 0 AROMATIC # 9 9 5 AROMATIC # 10 0 10 DATIVE # 11 1 10 DATIVE # 12 2 10 DATIVE # 13 3 10 DATIVE # 14 4 10 DATIVE # 15 5 10 DATIVE # 16 6 10 DATIVE # 17 7 10 DATIVE # 18 8 10 DATIVE # 19 9 10 DATIVE
シクロペンタジエニルアニオンは全てAROMATIC、Feとの間はDATIVE結合となったオブジェクトを作成することができました!!
これをSMILESおよびCXSMILESに変換してみましょう。まずはSMILES。
Fer_2_smi = Chem.MolToSmiles(Fer_2_RW) print(Fer_2_smi) # c12->[Fe+2]3456789(<-c1c->3[cH-]->4c->52)<-c1c->6c->7[cH-]->8c->91
The RDKit Book SMARTS Support and Extensionsの項の記載によると、RDKitではSMILESの拡張として配位結合を<-、->で表せるようになっているそうです。
この配位結合の表現と芳香属性炭素(c)の表現が組み合わさったSMILESになっているようです。
つづいてCXSMILESです。
Fer_2_CXSMILES = Chem.MolToCXSmiles(Fer_2_RW) print(Fer_2_CXSMILES) # c12->[Fe+2]3456789(<-c1c->3[cH-]->4c->52)<-c1c->6c->7[cH-]->8c->91 |^2:1|
先にみたように、配位結合はMolToCXSmilesで書き出せるプロパティの中に入っていません。
ですのでSMILES文字列のあとの特徴には配位結合の情報が含まれていないようです。
追記されている特徴^2:1はラジカルを表す表現のようです。ここではAtom Index 1のFeが2価のラジカルとして認識されています。確認のためCXSMILESからMolオブジェクトを作成し、Atom Indexと共に描画してみましょう。
mol = Chem.MolFromSmiles(Fer_2_CXSMILES) d2d = rdMolDraw2D.MolDraw2DSVG(200,200) d2d.drawOptions().addAtomIndices=True d2d.DrawMolecule(mol) d2d.FinishDrawing() SVG(d2d.GetDrawingText())

Atom Index 1のFe2+に対してシクロペンタジエニルアニオンから配位結合がでていることが無事確認できました。
CookBookのやり方
と、ここまで書いてきてRDKit CookBookにOrganometallics with Dative Bondsという項目があることに気づきました。そのまんまやん!
CookBookで書かれている方法を参考にGetNeighbors()をつかって配位結合を設定してみたいと思います。
Fer_3 = Chem.MolFromSmiles('c12c3c4c5c1[Fe]23451234c5c1c2c3c45', sanitize=False) # RWMolに変換 rw_mol = Chem.RWMol(Fer_3) # Fe(原子番号26)のAtomを取得 for atm in rw_mol.GetAtoms(): if atm.GetAtomicNum() == 26: metal = atm # Feの近接原子を取得 nbrs = metal.GetNeighbors() # Feとの結合をDativeBondに書き換える for nbr in nbrs: rw_mol.RemoveBond(nbr.GetIdx(), metal.GetIdx()) rw_mol.AddBond(nbr.GetIdx(), metal.GetIdx(), Chem.BondType.DATIVE) rw_mol # エラーは省略

print(Chem.MolToSmiles(rw_mol)) # c12->[Fe]3456789(<-c1c->3c->4c->52)<-c1c->6c->7c->8c->91
先の2例目と異なりシクロペンタジエニルアニオンのアニオンを設定していないので、Kekule化できないエラーはそのままですが配位結合は設定できているようです。
気づかぬ間にRDKit CookBook内容がすごい更新されてました。はじめからこっち見ておけばよかった。。。
まとめ
以上、今回はSMILESの拡張表現の一つ、CXSMILESとその応用としてFerroceneをどのようにSMIELSで表せるか、という問題にアプローチしてみました。
特徴の追記ができることで、線形表現のままより複雑な情報が表せるというのはとても便利そうです。
一方で、副次的ではありますがChemAxon社のページで記載されていた小文字のcを用いたフェロセンの表記([Fe].c1cccc1.c1cccc1)がRDKitではエラーが出てしまうこと、ソフトウェアによるSMILES表記の取り扱いの違いがわかった点で興味深いと思いました。
適当なことをあれこれ書いているので間違い等ご指摘いただければ幸いです。
*1:以下の記載はChemAxson社ページの記載を引用しています
*2:RDKit Bookの内容をCC BY 4.0のライセンスのもとに利用させて頂いています。
*3:ChemAxsonのドキュメントではSMILESに記載されていますがおそらくSMARTの方が正しいと思います。「化学の新しいカタチ」さんの記事 SMILES記法は化学構造の線形表記法がとても参考になります。
*4:参考:化学の新しいカタチ「RDKitの分子Molオブジェクトを扱う」
*5:RDKitドキュメント Getting Started In Python
HELMで核酸医薬をお絵描き
前2回の記事でマクロ分子の線形表現HELMについて概要と利用ツールをみてきました。
「いろいろなモダリティの表現に良さそう!!」ということですが、実際どんな感じになるのでしょうか??
今回は核酸医薬の具体例をお絵描きしていきたいと思います。
核酸医薬の概要
といっても私は核酸医薬をよくわかっていません。
国立医薬品食品衛生研究所 遺伝子医薬部 井上貴雄先生の以下2件の解説に特徴と現状がまとまっており非常にわかりやすかったです。(いずれもオープンアクセス)
- 文献1) 井上貴雄・佐々木澄美・吉田徳幸 核酸医薬開発の現状と今後の展望 Drug Delivery System 2019(34)86-98
- 文献2) 井上貴雄 核酸医薬 〜低分子、抗体に続く第3のモダリティ〜 SAR News No.38 (Apr 2020) 1-8
以下、引用させていただきつつ、基本的な内容を辿りたいと思います。
長くなってしまったのでご存知の方は飛ばしてください。
核酸医薬の特徴
まず、核酸医薬品とは一般に以下のような医薬品を指します。
上記の特徴は、核酸医薬と遺伝子治療用製品との違いを踏まえると納得しやすいかもしれません。(後者は翻訳されたタンパク質が作用(②)し、生物学的に製造される(③))
さて、従来の医薬品(低分子、抗体)と比較した核酸医薬の特徴としては、以下3点が挙げられています。
加えて、新規モダリティとして核酸医薬には従来の低分子と抗体の利点を併せ持つことが期待されています。具体的には以下2点。
- 抗体同様、高い特異性と有効性をもつ
- 低分子同様、化学合成で製造可能できる
(2.の利点はより具体的には低コスト、高均質性でしょうか??)
また、核酸医薬は一度開発スキームができれば、異なる創薬標的に対しても迅速に開発可能なことが期待されます。理由は以下3点。
核酸医薬の課題
このように魅力的な特徴のある核酸医薬品ですが、薬効本体であるオリゴ核酸に由来する、生体内での安定性・有効性という課題がありました。
これらの課題に対して修飾核酸技術や薬物送達技術(DDS)といった技術発展により改善が見られ、 現在の全身投与でも高い効果を発揮する候補品の開発に至っている、とのことです。
核酸医薬の薬物送達技術
核酸医薬の実用化に寄与した技術のうち薬物送達技術にはどのようなものがあるでしょうか?
臨床応用されているデリバリー技術は大きく①Lipid Based Nanoparticle、②Polymer Based Nanoparticle、③Conjugated Delivery Sytemに分かれるそうです。 *1
文献2 表1にはPEG、GalNac、LNPという例が記載されています。*2
| 薬物送達技術 | 商品名 | 一般名 | 分類 | 適応 | 投与 |
|---|---|---|---|---|---|
| PEG | Macugen®︎ | pegaptanib | アプタマー | 滲出型加齢黄斑変性症 | 硝子体内投与 |
| GalNAc | Givlaari®︎ | givosiran | siRNA | 急性肝性プリフィリン症 | 皮下投与 |
| LNP | Onpattro®︎ | patisiran | siRNA | 遺伝性ATTRアミロイドーシス | 静脈内投与 |
前2例(PEG、GalNac)は核酸そのものに薬物送達のためのユニットを直接(共有)結合させるもので、 後1例(LNP)は脂質ナノ粒子(Lipid Nano Particle)という粒子に核酸を内包させるDDS技術です。

3剤の内、全身投与という観点ではgivosiran、patisiranが当てはまります。 これら2剤はどちらもAlnylam社によるもので標的臓器は肝臓となっています。
例えばgivosiranの場合、GalNAcリガンドは上図のような構造となっているそうです。*3
末端に結合するN-アセチルガラクトサミン(GalNAc)は肝実質細胞の細胞表面に発現するアシアロ糖タンパク質(asialoglycoprotein receptor, ASGPR)に認識され結合します。 そして、受容体介在型エンドサイトーシス(receptor-mediated endocytosis)によって取り込まれることで、細胞内において核酸の効果が発揮されるとのことです。 *4

なぜhydroxyproline誘導体をかませているのか?炭素鎖の長さの影響は?GalNAcを3つぶら下げてるのは?などなど色々と興味深いところです。
脂質ナノ粒子
全身投与のもう一方patisiranは脂質ナノ粒子(LNP)によって体内動態がコントロールされています。
Alnylam社のOnpattro製品解説ページの図が非常にわかりやすいですが、 とても興味深い輸送メカニズムとなっています。
標的臓器、肝臓に到達するポイントは2点。
- 肝臓が大量の血液が潅流する臓器であること
- ApoE依存的機構
①は静脈内投与なので血液が集まりやすい肝臓に行きやすくなるのはなんとなくわかります。
面白いのは②で、肝臓分布過程でLNPに内因性アポリポタンパク質E(ApoE)が結合するそうです。 肝細胞表面の低比重リポタンパク質受容体(LDLR)などのApoE結合受容体を介して細胞内に取り込まれる、とのこと。
Alnylam社の2010年の文献ではapoE-/-マウス、LDLR-/-マウスを用いた解析が行われています。*5
中性リポソーム(neutral liposome)にアポリポタンパク質が吸着することが一般に知られていて、 その中でアポリポタンパク質Eだけが肝細胞への取り込みを促進できるから、ということに基づく研究のようです。
・・・すごい。
中性と書きましたが、ここも大事なポイントです。
実験室レベルでは核酸送達のキャリアとしてカチオン性キャリアがよく用いられるそうです。*6
カチオン性を用いることの利点は2つ
一方で核酸医薬のDDSに応用するにあたって困難な問題点も指摘されています。
- 細胞毒性:細胞内タンパク質との非特異的吸着による阻害
- キャリア正電荷により核酸が細胞内でもリリースされづらい
- 生体内組織への非特異的吸着により初回通過臓器(肝臓、肺)に捕捉されやすい
- in vivoで血中の細胞、タンパク質との非特異的結合により凝集、不活化、排出される
そこで臨床応用にあたっては非カチオン性キャリアが重要になってきます。 特にLNPでは非カチオン性脂質とpH感受性カチオン性脂質のハイブリッドといったアプローチがとられています。
このpH感受性というのがとても面白いポイントで、安全性や核酸輸送にとって以下のような利点があるそうです。*7
- 血中pH7.4ではほとんどイオン化しない(中性)ので、細胞障害性が低い
- 細胞内に取り込まれたのち、エンドソーム・リソソーム内の弱酸性pHでイオン化
- イオン化するとエンドソーム・リソソーム膜と融合し、内包核酸を細胞質に効率よく放出
patisiranで用いられているpH感受性カチオン性脂質はDLin-MC3-DMAというもので、pKa6.5前後、3級アミンを有する構造です。

このLNP技術は元々Pieter Cullis 教授らにより2010年 Nature Biotechnolgyに報告された下記文献が鍵となっているようです。*8 LNPの要素として1,2-dilinoleyloxy-3-dimethylaminopropane (DLinDMA)構造に基づく脂質のスクリーニングを行なっています。(読めなかった)
さらに2012年、Angewante Chemie Internal Editionに報告された文献では、 種々の親水性head groupを有する脂質を用いたpKaとin vivoでの効果の相関を調べており、 pKa 6.2-6.5が最適な範囲であるとしています。(オープンアクセス。Abstractの図がとても印象的です。)*9
Jayaraman M. et. al., Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 2012;51:8529–8533. https://onlinelibrary.wiley.com/doi/full/10.1002/anie.201203263
こういう化学構造のデザインで好ましい性質を達成していくのとても格好いいですよね。*10
修飾核酸
こちらは核酸医薬自体の安定性に関与し、具体的には生体内に存在する核酸分解酵素(ヌクレアーゼ)等による分解への抵抗性や、 標的との結合安定性の強化といった効果が期待されます。
文献2 SAR News No.38 の図2がわかりやすので以下に引用させていただきます。

まずリン酸部位の修飾です。ここには酸素原子を硫黄原子に置換したホスホロチオアート修飾(S化)がよく用いられます(S化オリゴ核酸、Sオリゴ)。
これにより以下の利点が期待されます。
- タンパク質との結合の向上、脂溶性の増加により細胞膜上のタンパク質を介した細胞親和性、膜透過性が向上
- 血中タンパク質との結合による血中滞留性の増加
- ヌクレアーゼ耐性の付与による整体安定性の改善
化学的な注意点として、リン原子上に不斉点が発生するためSオリゴは立体異性体混合物になってしまいます。
この立体配置を厳密に制御する技術を持った企業もあるそうです(Wave Life Sciences)。
文献*11によるとリン原子に不斉補助基を結合させたモノマーを使って、伸長・キラルS化を行なっているようです。*12
・・・すごい。
続いて糖部の修飾です。これによりRNAとの結合力の強化が期待され、2'位の修飾と架橋型修飾が例としてあげられています。
2'位の修飾としては上図に引用した2'-MOE以外にもフッ素化体(2'-F)、メトキシ体(2'-OMe)などがあります。
架橋型は糖部の2'と4'を架橋することでゆらぎのある糖部の立体配座を固定化します。これにより以下が期待できます。
- 相補鎖との結合力の向上
- 立体障害によるヌクレアーゼ耐性
他にも糖部修飾そのものではありませんが糖部にモルフォリノ環を用いたモルフォリノ核酸が知られており、
- ヌクレアーゼで分解されない
- 毒性が低い
といった利点があります。
以上が、修飾核酸の概要でした。 こういった様々な非天然のユニットが活用されていることが、HELMにおけるモノマーの取り扱いの柔軟さとどのように結びつくか、気になるところです。
核酸医薬の分類
核酸医薬の構造的特徴をざっとみたので、最後に分類についても触れておきます。
文献2 SAR News No.38 の表2がわかり易いので引用させていただきます。

この分類は「RNAを標的とするか、タンパク質を標的とするか」でとらえるとわかり易い、とのこと。
表2の左3つ(アンチセンス、siRNA、miRNA)がRNAを標的、右3つ(デコイ、アプタマー、CpGオリゴ)がタンパク質標的に相当します。
RNAを標的とする中でもアンチセンスは病因となるタンパク質を減少させたり、機能的なタンパク質を増加させたりと様々な作用機序があるそうです。
ちなみに薬物送達技術のところで取り上げた3剤はそれぞれ、① Macugen®︎(pegaptanib) アプタマー、② Givlaari®︎(givosiran) siRNA、③ Onpattro®︎(patisiran) siRNA となっていました。
分類は以上です。
以上、核酸医薬の基本的な事項について辿ってみました。
面白い話題が多くてどんどん脱線してしまいましたが、構造の複雑さと背景を知るのが表現方法を考える上で大事ということで。。。
HELM表現と他の表現の比較
では漸くですがHELM表記を試してみましょう。
文献2 SAR News No.38 の表1に記載されている中から化学修飾の異なる以下の3つを試してみたいと思います。

Exsondys®︎ (eteplirsen)
まずはSarepta TherapeuticsのExsondys®︎ (eteplirsen)から。
Drugs@FDAで閲覧できるLabel(NDA 206488)記載の構造はこんな感じです。

えらいごっついですわ。。。
表現の比較対象としてIUPHAR/BPS Guide to Pharamacologyの2D表現を引用してみます。 Guide to Pharmacologyは専門家によりキュレーションされたリガンド-活性-標的関係についてのデータベースで、CC BY 4.0で利用可能です。
Guide to Pharamacology; eteplirsenの核酸配列の2D表現はこんな感じ。

配列はわかりやすいですが、この表現ではモルフォリノ核酸であることやリン酸の修飾といった情報が表現できていないようです。
ではHELM editorをつかって構造を描いていきましょう。
まずはmonomerの確認してみます。モルフォリノ核酸糖部はあるようです。

リン酸部位と5'末端の構造は無いようなのでMonomer Managerに登録しましょう。
前者はPolymer Type RNA、後者はCHEMとしました。

まずは天然の核酸の場合のHELMを確認します。HELM Web-editorがお手軽です。
配列を「CTCCAACATCAAGGAAGATGGCATTTCTAG」をSequenceに入れてApplyするとグラフ表現とHELMが確認できます。

糖部位はすべてモルフォリノ核酸なので[mph]で、リン酸部位は新しく作ったモノマー[DMP]で置き換えていきます。
複数文字の略記なので角括弧[]で囲むことに注意します。
Pythonのreplaceで文字列置換してみます。RではなくR(を[mph](で置換しているのはRNAのRも置換されてしまうからです。
natural_analogue = 'RNA1{R(C)P.R(T)P.R(C)P.R(C)P.R(A)P.R(A)P.R(C)P.R(A)P.R(T)P.R(C)P.R(A)P.R(A)P.R(G)P.R(G)P.R(A)P.R(A)P.R(G)P.R(A)P.R(T)P.R(G)P.R(G)P.R(C)P.R(A)P.R(T)P.R(T)P.R(T)P.R(C)P.R(T)P.R(A)P.R(G)}$$$$'
mph_analogue = natural_analogue.replace('R(', '[mph](')
DMP_analogue = mph_analogue.replace(')P', ')[DMP]')
print(DMP_analogue)
# RNA1{[mph](C)[DMP].[mph](T)[DMP].[mph](C)[DMP].[mph](C)[DMP].[mph](A)[DMP].[mph](A)[DMP].[mph](C)[DMP].[mph](A)[DMP].[mph](T)[DMP].[mph](C)[DMP].[mph](A)[DMP].[mph](A)[DMP].[mph](G)[DMP].[mph](G)[DMP].[mph](A)[DMP].[mph](A)[DMP].[mph](G)[DMP].[mph](A)[DMP].[mph](T)[DMP].[mph](G)[DMP].[mph](G)[DMP].[mph](C)[DMP].[mph](A)[DMP].[mph](T)[DMP].[mph](T)[DMP].[mph](T)[DMP].[mph](C)[DMP].[mph](T)[DMP].[mph](A)[DMP].[mph](G)}$$$$
できました!お手軽!
HELM Editorで読み込んでみます。

グラフ表現でました!小さいので5'末端の拡大図も載せました。
ここに5'末端の修飾を足します。リン酸の追加とChemical Modifierから先に作成したモノマーを選んで貼り付け。
こんな感じになりました。

先の「Guide to Pharmacology」のような2D表現と比較してどうでしょうか?
HELMのグラフ表現ではモノマーが拡張可能かつ塩基、糖、リン酸の3つに分かれており、非天然の構造修飾をもった核酸であることが明確に示されています。
また、モノマーライブラリを介してより具体的な構造式と結びついているため、このグラフ表現は実際の構造式の情報と紐づけることが可能です。
一方で、構造式そのものよりも単純化されているため塩基の配列といった情報を把握しやすいものともなっています。
ではこのグラフ表記のHELMはどのようになっているでしょうか?
作成したグラフ表現の他の表記はEdit のViewから確認できます。

HELM表記の変更点はSimple polymer listの最初とConnection listです。 「RNA1{[DMP]. ~~~~ |CHEM1{[EPR_5']}$RNA1,CHEM1,1:R1-1:R1$$$」という表現になっています。
SMILESやMolファイルといった表記も確認できます。線形表記同士、SMILESとHELMを並べてみましょう。

HELMはモノマーの略記(ID)が使えることで簡略化されています。
SMILESで途中でてくる「%xx (xは数字)」は2桁の番号を使って閉環位置を示すためのものだそうです。*13
・・・初めて見ました。
このSMILESが他のソフトでも認識できるものなのか?念のためRDKitで構造を起こしみます。
from rdkit import rdBase, Chem from rdkit.Chem import Draw from rdkit.Chem.Draw import IPythonConsole # smi = SMILES文字列長いので省略 m = Chem.MolFromSmiles(smi) m

できました!わけがわからないがMolオブジェクトはできた!
Spinraza®︎ (nusinersen)
つづいてSpinraza®︎(nusinersen)です。Novaltis社のZolgensma®︎(Onasemnogene abeparvovec-xioi)との関係でも話題となりましたね。
構造は以下の通りです。

特徴としてはリン酸がS化されたSオリゴであることと、糖が2'-MOEとなっていることです。
こちらはPubChemにHELM含めて色々な表現が載っていました!こんな感じ・・・

他にもInChiやCanonical SMILES、Isomeric SMILESが載っていました。情報が載っているものと見当たらないものの違いは何なのでしょう??
HELM editorに入れる前にモノマーがあるかを確認しておきます。

デフォルトで必要なモノマーが揃っていそうです。モノマーライブラリの追加が必要ないのでHELM Web-editorで描画してみます。
HELMをはりつけてApply!!

HELMがPubChem等で提供されているとお手軽ですね!
HELM表記およびグラフ表現では修飾構造(5meC、sP、MOE)といった点がわかりやすいように思います。
Givlaari®︎(givosiran)
最後、Givlaari®︎(givosiran)です。

先の2つよりも構造の特徴が多いようです。
- リン酸部位にO体とS体の両方
- 糖部位は2'-F体と2'-OMe体が使われている
- 2本鎖
- GalNAcコンジュゲート結合
まずはモノマーの確認です。2'-Fと2'-OMeはあるようです。

コンジュゲートを用意しましょう。こんな感じで登録してみました。

ある程度HELMの書き方がわかってきたのでStrandごとのSimple Polymerを書いてみます。

HELM editorに描いていきます。StrandごとにHELMを読み込んでいきましょう。

Sense StrandとAntisense Strandをどちらも読み込むと図のようになります。 どちらも左から右に向かって5'-3'の向きとなっています。
この状態で図の上にあるHybridizeボタンを押してやります。
するとAntisense Strandの左右がひっくり返り、2つのStrand間に水素結合が形成されます。

拡大してみます。塩基の間のドットが水素結合を表しています。

この状態でHELMは以下のような表現となっています。
水素結合がpairとして表されています。

最後にsense鎖(ss)、antisens鎖(as)を明確にするためAttribute Listに属性を付け加えることもできます。こんな感じ、、、

以上、givosiranのHELM表記とグラフ構造でした。
givosiranは添付文書の構造式の書き方がかなり分かりやすいのでHELMにしたことで可読性がよくなったか?というとちょっと微妙です。
ですが、モノマーライブラリと紐づいているので、略語の説明を個別にする必要がなく、糖、塩基の結合位置も構造式レベルで明確に指定されている、という点では HELMを使うことに利点があるのかもしれません。
またGalNAcコンジュゲートを一つのCHEMモノマーとして登録しましたが、リンカーと糖(GalNac)を別々に登録すれば、 異なる糖の組み合わせのコンジュゲートライブラリを表現し、それぞれの性質と結びつけることもできそうです。
まとめと感想
以上、今回は核酸のHELM表現を試してみました。
実際に使われている医薬品でHELM表現を試すことでいくつかメリットを感じられたように思います。
- 異なるタイプの核酸医薬に対して同じモノマーライブラリに基づいた共通の表現ができる
- 実際の構造式に紐づいており、修飾構造を明確に表現できる
- IDで表記するのでユニットの文字列置換で簡単に記述を変更できる
と、いったところでしょうか?
それぞれ単独の構造のみで記載したのであまり"旨み"がなかった感もありますが、 これらの裏側にあるであろう大量の核酸医薬候補ライブラリーを表現、管理、データベース化する、 となった場合に中々使い勝手の良いものとなるのではないでしょうか???
一方で、使いにくさを感じる点としては、コンジュゲートのような化学修飾ユニットをCHEMモノマーとしてそれぞれ登録する必要があることです。
化学修飾はSMILESで書いてハイブリッドな表現ができたらもっと便利な気もしますが、やはり難しいのでしょう。
あとPuChemやChEMBL、思ったより欲しいHELM表記あまり載ってなかったです・・・。 もう少し核酸医薬分野のデータベースも手厚くして欲しい気もします・・・が、ひょっとしたらHELMの標準ライブラリ、IDに修飾核酸をどこまで含めるか?みたいな問題があるのかもしれません。
毎度のごとく知識不足すぎて基礎事項を調べていたら本題のHELM表記に中々たどり着けませんでした。。。
まあ色々面白かったのでいいや。
以上!
今回も色々と間違いが多そうです。ご指摘いただければ幸いです。
*1:久保田恒平博士 博士論文「siRNA 担持脂質ナノ製剤の評価 に関する研究」記載より CiNii
*2:下表は文献2 表1より抜粋・改変して作成
*3:Pharmaceuticals 2020, 13(3), 40 CC BY 4.0の元に図を引用
*4:Toxicologic Phatology 2018, 46, 735 CC BY 4.0の元に図を引用
*7:菊池 寛 Drug Delivery System 2019(34)106 企業的観点からみた核酸・遺伝子医薬品開発の現状と今後の展望:DDSとの関わり
*8:Semple, S. et. al., Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 28, 172–176 (2010).
*9:Jayaraman M. et. al., Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 2012;51:8529–8533.
*10:こちらのレビューの図も良いです Mol Ther. 2017(25)1467
*11:Nat. Biotecnol. 2017(35)845