コンフォメーションとRadius of gyrationによる評価について
巨大なPROTACがなぜ細胞膜を透過するのか?溶液中におけるコンフォメーションの違いから解析した興味深い文献を読みました。
Solution Conformations Shed Light on PROTAC Cell Permeability
Yoseph Atilaw, Vasanthanathan Poongavanam, Caroline Svensson Nilsson, Duy Nguyen, Anja Giese, Daniel Meibom, Mate Erdelyi, and Jan Kihlberg ACS Medicinal Chemistry Letters Article ASAP DOI: 10.1021/acsmedchemlett.0c00556 (https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00556)
CC-BY 4.0の元で公開されています。
この論文中で出てきた Radius of gyration (Rgyr) というものがよくわからなかったので調べてみました。
合わせて、この記事では以下の内容を扱っています。
- PDFファイルからのテキスト情報抽出
- 化学情報のファイル形式xyz形式とその変換
- 論文の概要
- Radius of gyration (Rgyr)
- PDFから座標を抜き出す
- 座標のXYZ形式とPyMolでの確認
- Open Babelによるファイル形式の変換と修正
- RDKitによるRadius of gyrationの計算
- PyMolでradius of gyrationを描画
- まとめ
論文の概要
まずは論文の中身についてざっと紹介します。
PROTACは概して分子量が大きく、LipinskiのRule of 5からは大きく外れた構造をとっています。このような巨大な分子が細胞膜を透過する、経口投与で薬効を示す、というのは非常に驚きです。
では何故膜透過が可能なのでしょうか?
その一つの解として、こちらの論文では、溶液中でのコンフォメーションの違いが寄与しているのではないか?と示唆しています。つまり細胞膜中のような疎水性環境下では、丸まったコンフォメーションをとっており、実質的な体積(大きさ)は小さくなっているのではないか?、ということのようです。
解析されている構造例は以下。グラフィカルアブストラクトがわかりやすいですね。
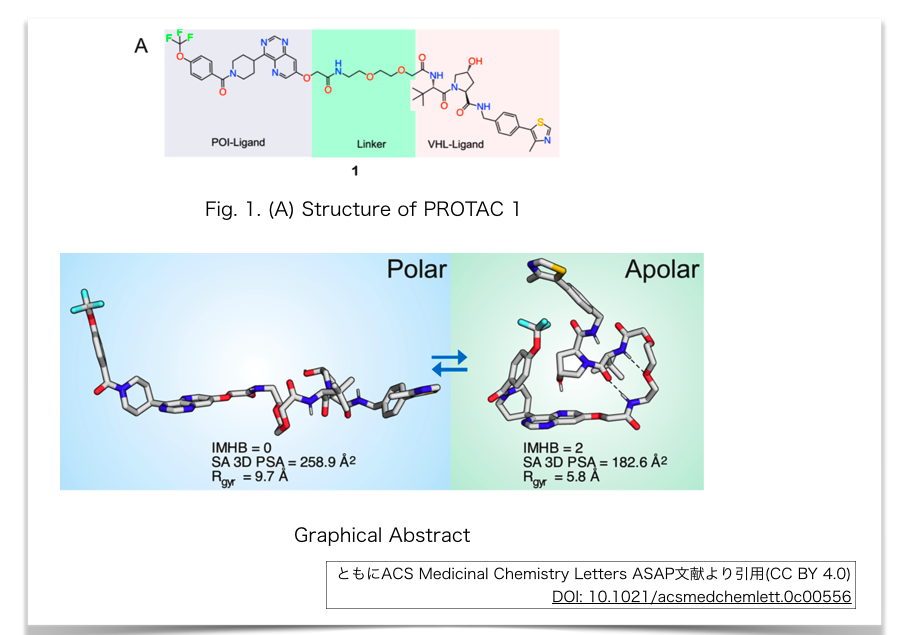
以下の解析手法でコンフォメーションを求めているようです。
- コンフォメーション解析の元となる実験データはNMR
- NMRデータ解析手法はNAMFIS(NMR analysis of molecular flexibility in solution)アルゴリズム
- NAMFISでは様々なコンフォメーションの時間平均であるNMRデータを、各コンフォーメションの占有率に分解して解析可能 *1
- NAMFISに必要なコンフォメーションのアンサンブルはMCMM (Monte Carlo molecular mechanics) conformational searchで生成*2
得られたコンフォメーションを評価する指標として、以下を用いています。
SA 3D PSAの具体的な計算方法は理解できていませんが、化学の新しいカタチさんの記事「トポロジカル極性表面積は計算コストの低い推定PSA」が参考になると思います。
PSAはオクタノール/水分配比率(logP)などと同様に、分子の極性・脂溶性を示す記述子になります。PSAと膜透過性に関する実験データとの間にはよい相関があることが判明しています。例えばあまりに大きなPSA(140 Å2以上)を有する化合物は膜透過性が低いことが経験的に知られています。(化学の新しいカタチ 上記記事より引用)
得られたコンフォメーション解析結果から、PROTAC 1は疎水性環境中では丸まった構造をとっていることが示唆されました。
このコンフォメーションを安定化させる構造的特徴として以下が指摘されています。
- 分子内水素結合(IMHB、intramolecular hydrogen bond)
- 芳香族へテロ環(チアゾール、ピリミジン)間のπ-π interaction
- ヒドロキシ基とピリミジン間の非古典的水素結合(nonclasical hydrogen bond)
IMHBやnonclasical hydrogen bondは、アミドNHやヒドロキシ基を溶媒接触面からマスクすることでもSA 3D PSAを低下させ、膜透過の向上に関与しているのではないか?ということが指摘されています。
この解析結果が非常に興味深いと思ったのは、一見すると2つの大事なユニットをつなぐだけに見えるflexibleなPEGリンカーが、分子内水素結合という形で3次元構造の安定化に関わり、膜透過の向上という物性変化に積極的に寄与している可能性が示唆されている点です。
回転可能な結合数(Nubmer of rotatablebond、NRotB)は極性表面積(PSA)と同様に、一般に膜透過と逆相関することが指摘されている指標のように思います。NRotBを増加させる自由なPEGリンカーが、実は膜透過に大きな役割を果たしているかもしれない、カメレオン的特性のキモ、というのはとても面白い示唆だと思います。
Radius of gyration (Rgyr)
で本題、Radius of gyrationとは何なのか?
Wikipediaによると断面回転半径と訳されるらしく、断面の性質を表すパラメータの一つだそうです。

何のこっちゃ????
英語版のWikipediaの方がもう少しわかりやすいです(Radius of gyration (wikipedia))。
曰く、「慣性モーメント(moment of inertia)を持つ点までの半径方向距離(radial distance)」です、と。
数学的には「重心、あるいは与えられた軸からの物体の各パーツまでの距離の二乗平均平方根」となるそうです。
・・・なるほどわからん。ポリマー科学におけるIUPACの定義も記載されているので、ポリマー界では一般的に使われる指標なのでしょうか???
Wikipediaの表式は軸(axis)についてのものっぽいです。重心(center of mass)についての式はこちらのページにありました。

Rgyrが小さいほど原子が重心に近く、大きいほど遠くなるそうです。つまりRgyrが小さい方が3次元的に小さい(丸まっている?)ということのようです。
とりあえず座標と質量があれば計算できるようです。
ほな、やってみましょうか!!
PDFから座標を抜き出す
先のACS Med. Chem. Lett.論文ではコンフォマーの座標がSupporting Informationで提供されています。・・・PDFで。。。
とりあえずpdfminer.sixを使えばテキストを抽出することができるそうです*3 。pdfminer.sixのドキュメントはこちらで、GitHubはこちらです。
pip install pdfminer.six
一緒にダウンロードされるpdf2txt.pyを使えば簡単らしいです。
私はプログラミングが苦手なので、以降Jupyter notebookで実行しています。
まずPython3でpythonファイルを実行するため、エクスクラメーションマーク(!)をつけて!python3としています。pdf2txt.pyはAnacondaのbinの中にインストールされているようなので相対Pathを使ってpythonファイルを指定しました。
入出力は以下の通りです。
- 入力ファイル : ACSからダウンロードしたSupporting Information (「ml0c00556_si_001.pdf」)
- 出力ファイル(
-o):「SI.txt」
引数--page-numbersを使うと、抜き出したいページを指定できます(ページ番号をスペースを挟んで並べる)が、今回は目的のページ数が多く面倒なのですべて処理します。
!python3 ../../.pyenv/versions/anaconda3-5.3.1/bin/pdf2txt.py -o SI.txt ml0c00556_si_001.pdf
無事「SI.txt」ファイルができました。該当箇所はこんな感じ。
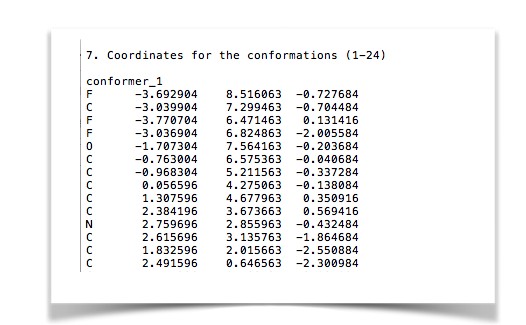
ただし、 ページ番号も同時に抽出されるので、座標の合間に改行やベージ番号が挟まっているためそのままでは使えません。 とりあえずコンフォマー1つ分座標があれば良いので手作業で取り除きました。
このままでもRgyrを計算することはできそうですが、折角ですので化学構造の取り扱いに適したファイル形式への変換を試みたいと思います。
座標のXYZ形式とPyMolでの確認
今回の座標の記載方法はXYZ形式に相当すると思います。化学の新しいカタチさんの記事「XYZ形式とZ-マトリックスは分子の立体構造を表す入力フォーマット」に詳しいのでご参照下さい。
上記記事によると、
という記載になっていれば良いようです。
論文の記載より今回の化合物1の組成式はC50H58N9O10F3S となっていますので原子の数は全部で131となります。 この情報を加えて「conformer_1.xyz」というファイルを作成しました。

xyzファイルが正しくできているか? 確認のためPyMolで描画してみます。
論文本文中のFig. 3とおおよそ同じ向きになるようにして並べてみました。
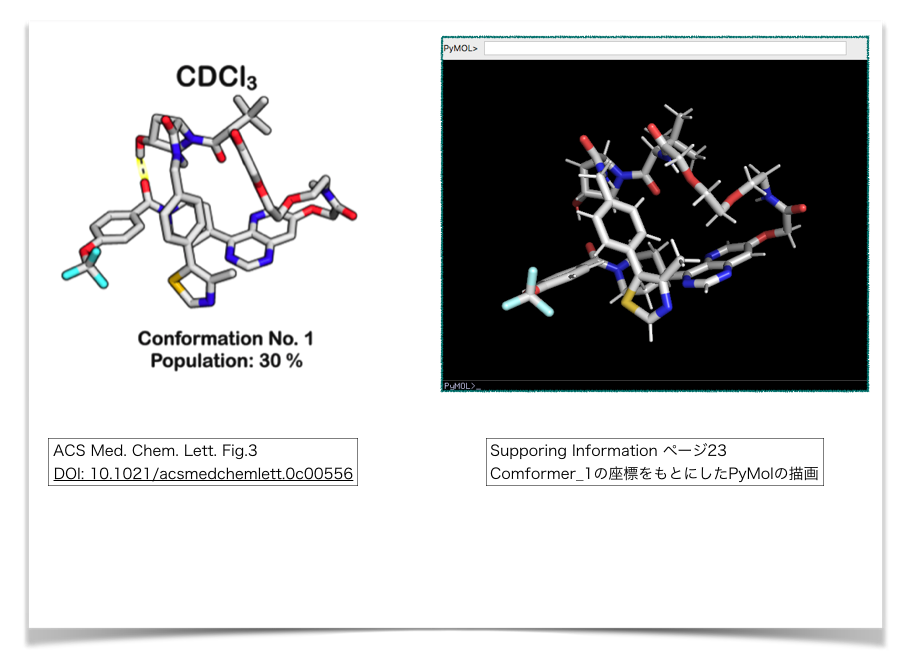
どうやらうまく情報を抽出できていそうです。
xyz形式は座標の情報しか入っていないようなので、結合次数の情報が無く、すべて単結合で表されているようです。・・・なるほど。
Open Babelによるファイル形式の変換と修正
欲しい情報を得られていそうなことがわかりました。ところで私はRDKitが好きなのですが、RDKitではxyzファイルを直接読み込むことができません。
そこで、Open Babelを用いてmolファイル形式に変換してみようと思います。ここではOpen Babelを扱えるPythonライブラリpybelを利用します。Pybelのドキュメントはこちらです。使い方は化学の新しいカタチさんの記事 「Open Babelを使って化学情報のファイル形式を変換」がとても参考になります。
扱えるファイル形式とその拡張子はドキュメントのこちら(Supported File Formats and Options)で確認できます。Jupyter notebook上でprint(pybel.informats)あるいはprint(pybel.outformats)として確認することもできます(入力 or 出力)。
早速、xyzファイルを読み込んでmolファイルに変換してみましょう!
readfile()でファイルを読み込めます。ただしこちらはジェネレーターオブジェクトです。今回は分子一つだけなので、__next__()で中身にアクセスして取り出しています。複数分子の時はforループで取り出せば良いらしいです。
import pybel single_obmol = pybel.readfile("xyz", "conformer_1.xyz").__next__()
出力はwrite()を使います。今回は1分子なのでそのままでOKです。
single_obmol.write('mol', './conformer_1.mol')
無事、conformer_1.molというファイルができました。
確認のためPyMolで描画してみましょう。先のxyzファイルの描画と並べてみます。
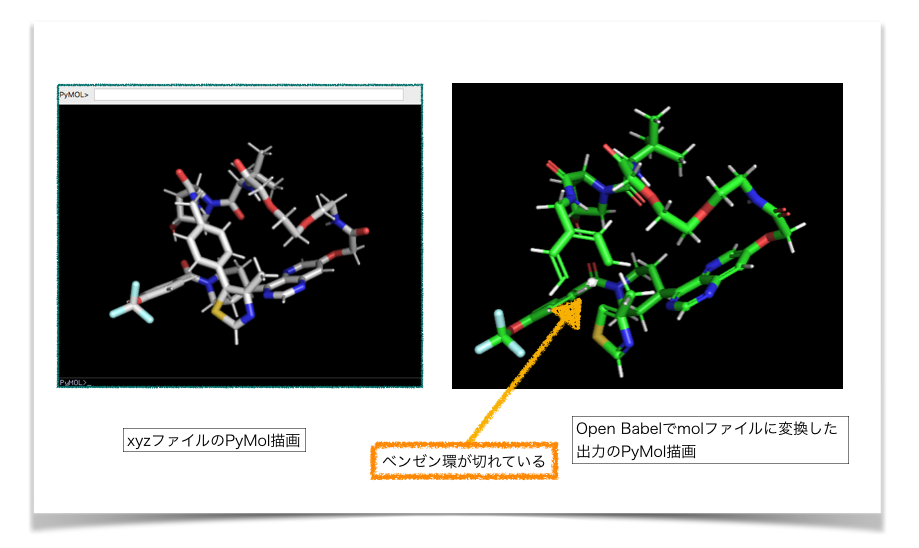
Open Babelで変換したMolファイルは単結合だけでなく2重結合も見えている一方で、ベンゼン環の結合が切れてしまっています。
どうしてこうなった??
おかしくなっている原子とその周りにAtom Identifiers: indexでラベルをつけてみました。*4。合わせてMolファイルの該当箇所を確認してみます。
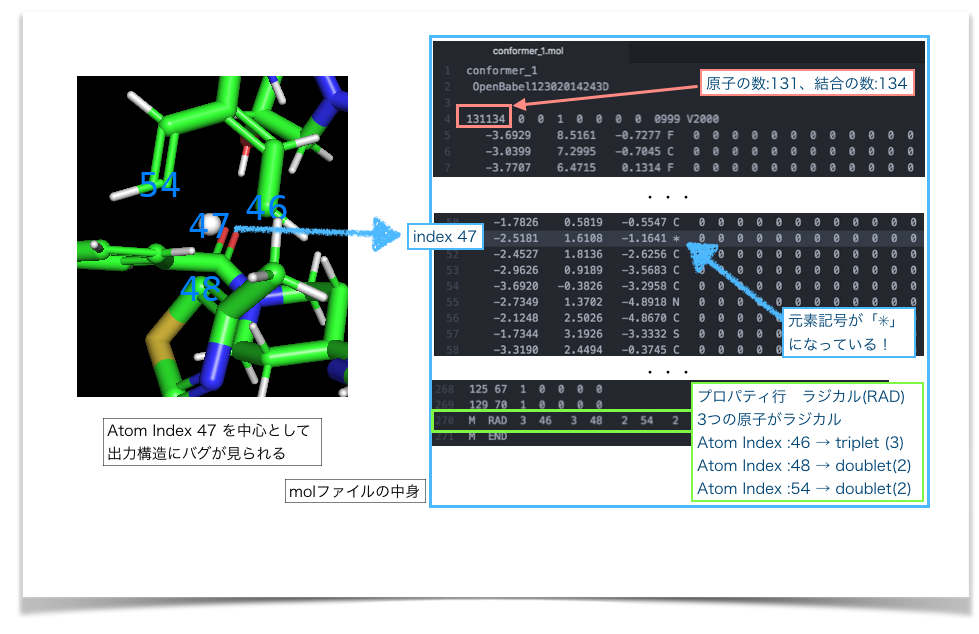
図のように、バグのある原子(Atom index: 47)は、molファイルのアトムリストで元素記号が「*」となっていました。また、結合リストでは47を中心とした結合関係が書かれていませんでした。結合次数が原子価に足りないAtom index 46、48、54はラジカルとして扱われてしまっています。
以上を踏まえて次のように修正してみました(conformer_1mod.mol)。
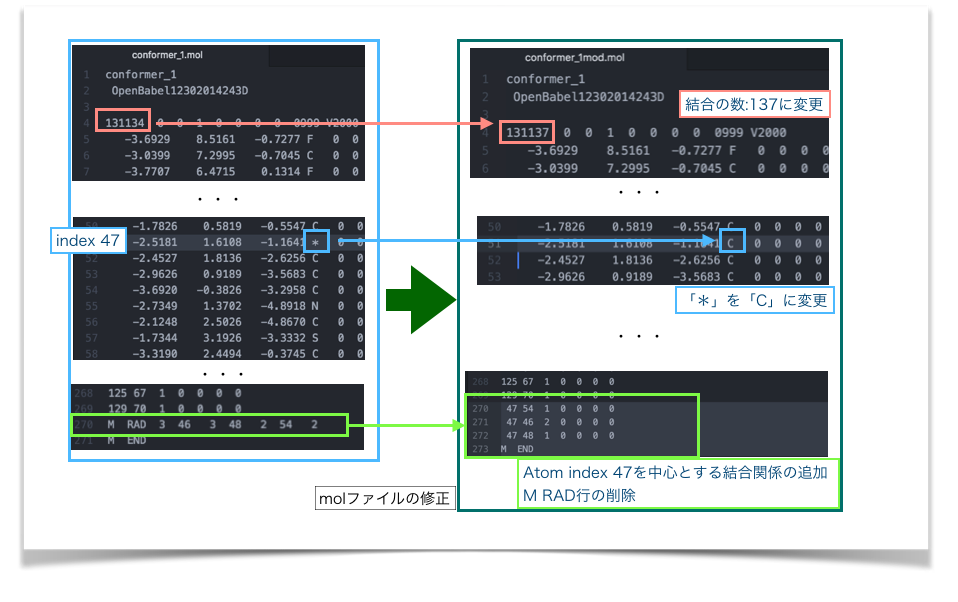
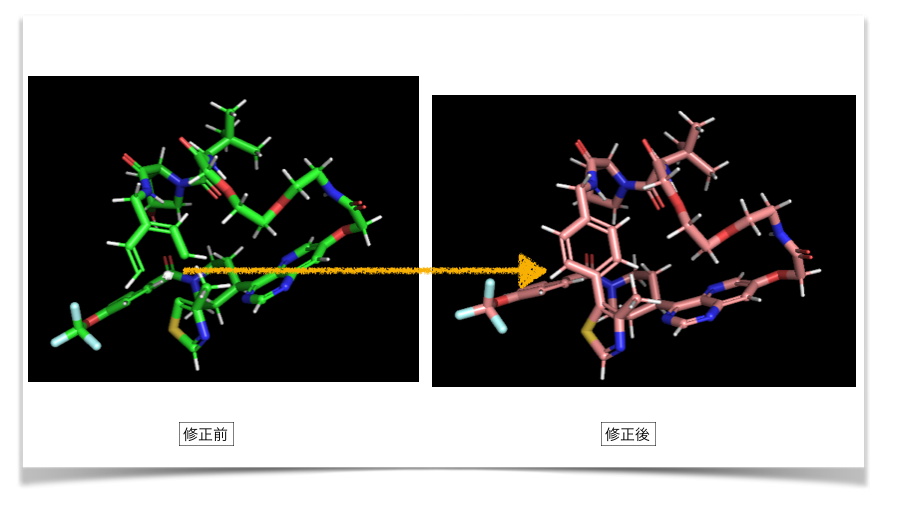
修正したmolファイルを再度PyMolで読み込み、修正前と比較してみました。
下図の通り、構造のバグが修正できているようです!!
修正したmolファイルがRDKitでも読み込めるか試してみましょう。
Molオブジェクトに変換したのち、py3DMolで3次元描画を行います。使い方は化学の新しいカタチさんの記事「py3Dmolを使って化学構造をJupyter上で美しく表示する」に詳しいです。
from rdkit import Chem from rdkit.Chem import AllChem, Draw from rdkit.Chem.Draw import IPythonConsole import py3Dmol conf1 = Chem.MolFromMolFile('./conformer_1mod.mol', removeHs=False) # 水素原子も必要 IPythonConsole.drawMol3D(conf1)
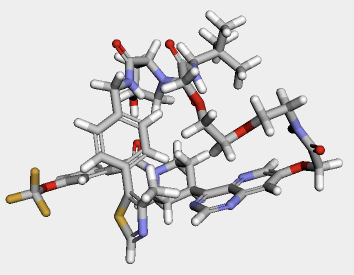
修正したmolファイルは無事RDKitで読み込むことができ、正しい構造を構築できていそうです。
ダメ押しで組成式も確認してみます。
from rdkit.Chem.rdMolDescriptors import CalcMolFormula formula = CalcMolFormula(conf1) print(formula) # C50H58F3N9O10S
Supporting Informationに記載のデータと同じ組成式が得られました。良かった良かった!!
なお、RDKitでxyzファイルを読み込めるようにするためのプログラムもあるようです。
- xyz2mol ( GitHub → https://github.com/jensengroup/xyz2mol)
- 使用例 Kaggle notebook → Using RDKit for Atomic Feature and Visualization
RDKitによるRadius of gyrationの計算
かなり遠回りしてしまいましたが、ここまでで論文からコンフォマーの座標の抽出と一般的な化学構造ファイルへの変換が終わりました。
本題、Radius of gyrationの計算を行ってみましょう!
PyMolWikiのRadius of gyrationを参考に、RDKitのMolオブジェクトから直接求められる形に修正を試みたいと思います。
まずは重心の座標(r_CM)を求めます。以下の手順でいいはず。。。
- Molオブジェクトから各原子の座標の取り出す
- 各原子の座標と原子量を掛け算して総和をとる
- 分子量で割り算
座標の取得は化学の新しいカタチさんの記事「RDKitによるコンフォマーの生成」を参考にさせていただきました。
こんな感じの関数を作ってみました。
def calc_r_CM(mol): from rdkit import Chem from rdkit.Chem import Descriptors import numpy as np # 各原子の原子量と座標を準備 mass_list = [] coordinate_list = [] mol_conf = mol.GetConformer(0) for i, (x, y, z) in enumerate(mol_conf.GetPositions()): # 原子量 atom = mol.GetAtomWithIdx(i) mass_list.append(atom.GetMass()) # 座標 coordinate_list.append((x,y,z)) # (x,y,z)それぞれに原子量x座標を計算し総和をとる r_CM_fraction = np.zeros(3) for m, (x, y, z) in zip(mass_list, coordinate_list): r_CM_fraction[0] += m*x r_CM_fraction[1] += m*y r_CM_fraction[2] += m*z # 分子量 MW = Descriptors.MolWt(mol) # 重心の座標 r_CM = r_CM_fraction / MW return r_CM
コンフォマー1のMolオブジェクトを投げ込んで重心の座標を求めてみます。
print(calc_r_CM(conf1)) # [-0.07220759 0.07596426 -0.02222654]
求まりました。ほぼ原点ですね。
続いて目的のRadius of gyration(RG)を求めます。先の関数と重複が多くなってしまいましたがとりあえずこんな感じ?
def calc_RG(mol): from rdkit import Chem from rdkit.Chem import Descriptors import numpy as np # 各原子の原子量と座標を準備 mass_list = [] coordinate_list = [] mol_conf = mol.GetConformer(0) for i, (x, y, z) in enumerate(mol_conf.GetPositions()): # 原子量 atom = mol.GetAtomWithIdx(i) mass_list.append(atom.GetMass()) # 座標 coordinate_list.append((x,y,z)) # 重心の座標 r_CM = calc_r_CM(mol) # 分母は分子量 MW = Descriptors.MolWt(mol) # 分子は(原子量 x (原子座標 - 重心の座標)^2 )の総和 r2_RG_fraction = 0 for m, (x, y, z) in zip(mass_list, coordinate_list): r2_RG_fraction += m * ((x - r_CM[0])**2 + (y - r_CM[1])**2 + (z - r_CM[2])**2) # 回転半径の2乗 r2_RG = r2_RG_fraction / MW # 回転半径 r_RG = np.sqrt(r2_RG) return r_RG
作成した関数でRadius of gyrationを求めてみます。
print(calc_RG(conf1)) # 5.481507768388569
約5.5Åと求まりました。さて正しい計算結果となっているでしょうか??
論文のFig.5 を引用します。
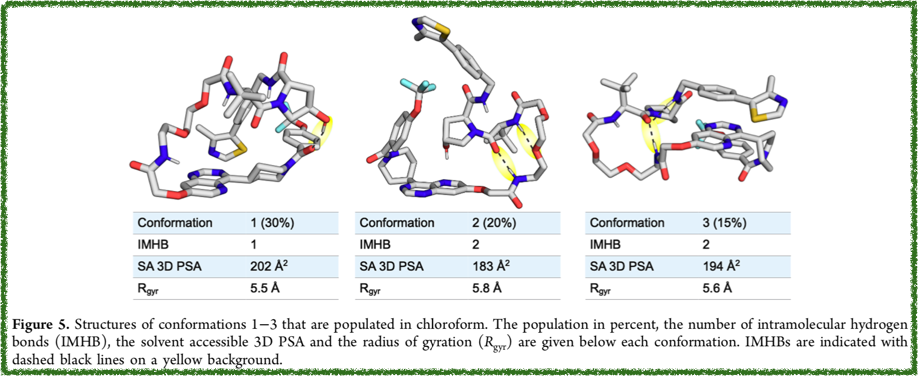
Conformation 1のRgyrは 5.5Åとなっています。どうやら合っていそうです。良かった!!
PyMolでradius of gyrationを描画
正しそうな結果は出ましたが、実際この数値の意味するものがうまくつかめません。
せっかく座標があるのでPyMolでコンフォマーと一緒に描画してみましょう!
Psudoatomを使えばいけそうです。
先に求めた重心の座標([-0.072, 0.076, -0.022] )とRgyr(5.48)を使って、PyMolのコマンドラインに以下を打ち込みました。
pseudoatom Rgyr, vdw=5.48, pos=[-0.072, 0.076, -0.022]
このままでは座標の点だけなのでShowでas meshとしてみました。

おおっ!!!
コンフォマーと重なるように球体が描かれました!!!!
確かにこれなら3次元的な構造を表す指標と言われても納得です!!!すげー!!!
まとめ
以上、今回は立体的な構造の指標、Radius of gyrationについて調べてみました。知らない指標だったのでPyMolWikiにも載っていてびっくりしました。
生物界隈でもよく使われる指標なのでしょうか??詳しい方教えていただけると幸いです。・・・あとNAFMISとSA 3D PSAについても教えてください。。。
また、実際に求める過程でxyz形式とその変換も試しました。予想外にOpen Babelで行った変換結果にはバグがありました。 ファイル形式の変換によく使われるソフトと聞いていたので、あれっ!!!ちゃうやん!!!って感じでした。
xyz形式は座標と原子の種類の情報しかないため、情報量が少ないことがバグが生じた原因なのかもしれません。
情報なんてなんぼ合ってもいいですからね!
いずれにせよファイル形式の変換は注意しないといけないということがわかって良かったです。
今回もいろいろと間違いが多そうなのでご指摘いただけると幸いです。
ではでは!
*1:どういうことかさっぱりわかりません。 ref. J. Am. Chem. Soc. 1995(117)1027
*2: Supportgint Informationより、ソフトウェアはシュレーディンガー社のMacromodelのようです
*3:【Python】pdfファイルからテキストを超簡単に抽出する方法
*4:PyMolのAtomラベルのサイズと色はPyMol Wiki:Labelを参考に変えています