JupyterNotebookでpymolのimportに失敗した話
PyMOL便利ですよね!でっかいタンパク質の描画も綺麗で動きも速いですし。
便利なPyMOLがCUIでも使えるらしいと知り、初心者の味方 jupyter notebook で使おうとしたところいろいろと躓いたので備忘録を残します。
まず結論!
- Pathを通して
ファイル名を変更「 _cmd.cpython-37m-darwin.so → _cmd.so 」
した!という話をだらだら書きます。
03/20追記
ファイル名変更よりも_cmd.cpython-37m-darwin.soのシンボリックリンク_cmd.soを作成する方が安全とのことです。
@Ag_smith先生ありがとうございました!
PyMOLの状態
PyMOLは @Ag_smith 先生の記事macOS/CentOS 7/Ubuntu 18.04へのオープンソース版PyMOLのインストール方法 にしたがってインストールしました。ちなみにパソコンはMacBookProです。
brew tap brewsci/bio brew install pymol
で、ターミナルでpymolと打ち込むとウィンドウが立ち上がることを確認しています。

Jupyter notebookでimport error
早速 import ! そしてエラーが出た!
import pymol --------------------------------------------------------------------------- ModuleNotFoundError Traceback (most recent call last) <ipython-input-1-f943b406a205> in <module> ----> 1 import pymol ModuleNotFoundError: No module named 'pymol'
「pymolなんて名前の奴はない」と怒られました。・・・あるよ。。。
PATHを確認
色々みているとPATHが通っていないのがよくないのでは、ということだったので、 まずはPyMOLがどこにインストールされているか確認して見ることにしました。 (コピペでインストールしてるからソフトの居場所も分からない。)
立ち上げたPyMOLのウィンドウで、コマンドを打ち込む枠(「PyMOL>」と書いてあるところ)に以下を貼り付けました。
import sys print(sys.path)
以下が表示されました。
['', '/usr/local/Cellar/pymol/2.3.0/libexec/lib/python3.7/site-packages', '/usr/local/Cellar/python/3.7.5/Frameworks/Python.framework/Versions/3.7/lib/python37.zip', '/usr/local/Cellar/python/3.7.5/Frameworks/Python.framework/Versions/3.7/lib/python3.7', '/usr/local/Cellar/python/3.7.5/Frameworks/Python.framework/Versions/3.7/lib/python3.7/lib-dynload', '/usr/local/lib/python3.7/site-packages']
HomeBrew経由でインストールしたものは「Cellar」というディレクトリの下に入るそうです。
どうも「'/usr/local/Cellar/pymol/2.3.0/libexec/lib/python3.7/site-packages'」が怪しそうです(pymolって書いてあるから)。
こちらを使うことにしましょう。
PATHを通すも新たなエラーが!
.bash_profile にPATHを追加すれば良いようですが、自信がないのでjupyter notebookでテストしてみます。
以下を打ち込み・・・
# PyMOLへのPATHを通す import sys sys.path.append('/usr/local/Cellar/pymol/2.3.0/libexec/lib/python3.7/site-packages') import pymol
新たなエラーを得た!
---------------------------------------------------------------------------
ImportError Traceback (most recent call last)
<ipython-input-3-f943b406a205> in <module>
----> 1 import pymol
/usr/local/Cellar/pymol/2.3.0/libexec/lib/python3.7/site-packages/pymol/__init__.py in <module>
581 # _cmd = sys.modules['pymol._cmd']
582
--> 583 from . import cmd
584
585 cmd._COb = None
/usr/local/Cellar/pymol/2.3.0/libexec/lib/python3.7/site-packages/pymol/cmd.py in <module>
102
103 import re
--> 104 from pymol import _cmd
105 import string
106 import threading
ImportError: cannot import name '_cmd'
今回は ModuleNotFoundError ではなかったので、先ほど追加したPATHで当たりだったようです。
PyMOLの居場所はわかったものの、今度は_cmdというのが見つからないらしい。
FinderでPyMOLのディレクトリを確認すると、似た名前の「cmd.py」というファイルはありましたが、「_cmd.py」は見当たりませんでした。 ひょっとして名前を付け間違えたか?と思い、「cmd.py」を複製し、「_cmd.py」にファイル名を変えてみましたが異なるエラーがでただけでした。
そんなアホなことでは直らないですよねー。アンダーバー、なんか意味ありそうですし・・・。
それっぽいファイルの名前を変えたら動いた!
色々と調べてもさっぱりわからず、もう一度PyMOLのディレクトリを見直すと怪しげなファイルがありました。
_cmd.cpython-37m-darwin.so
拡張子が「.py」でなく名前が長いので素通りしていましたが、最初に「_cmd」と書いてあります。 開いてみても文字化けしてさっぱり中身がわからないファイルでしたが、 「.so」というのはMacOSで使われる共有ライブラリの拡張子("shared object"の略)だそうです。*1
意味がわかりませんでしたが、とりあえず重要っぽいファイルでファイル名が探しているものに似ている。 ・・・名前変えちゃえ!ということで、ファイルを複製して邪魔っぽい部分(.cpython-37m-darwin)を消し、 「_cmd.so」にしてみました。
で、もう一度jupyter notebookに戻りimportすると今度はエラーを出さずに実行できました! エラーが出ないだけだと不安なのでPATHを確認してみます。
import pymol
pymol.__path__
['/usr/local/Cellar/pymol/2.3.0/libexec/lib/python3.7/site-packages/pymol'] と表示されました! どうやら通したPATHも合っていたようです!
まとめ
そもそもPyMOLのCUIでどのような機能が使えるかわかっていないので、importはできているっぽいものの本当に正しく使えるのか? ファイル名を変更してしまったけどよかったのか?等々不安が残ります。とりあえずの備忘録として残しておきます。
おそらく間違っているように思います。詳しい方ご教示いただければ幸いです。
結局 cpython-37m-darwin が何かよくわかりませんでした・・・
合成医薬品の変遷をバブルチャートで眺める話
あけましておめでとうございます(?) あれよあれよという間に2020年になりましたね。
オリンピックイヤーな今年はバブリーな一年にしたい!!・・・ということでバブルチャートで遊んでみました。
バブルチャートといえば、やはり書籍「ファクトフルネス」でも有名となったハンス・ロスリング博士らによる「動くバブルチャート」ではないでしょうか?
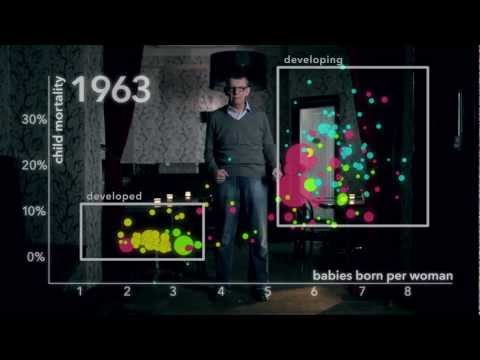
・・・こういうカッコいい図作りたい !!! と思ったら、好きなデータでグラフをかけるようにしてくださっている記事がありました。*1
そのライブラリ名は・・・Bubbly!!
バブリーさせていただきましょう!(訳:コピペした)
題材として、「医薬品として承認されている合成低分子が時代とともにどのように変化してきたか」眺めてみたいと思います。
ChEMBLからのデータ取得
まずはChEMBLからそれっぽいデータをとってきます。
トップページの「See all visualisations」をクリックすると、 「Drugs by Molecule Type and First Approval」というグラフがありました。 どうやら各医薬品分子が最初に承認された年を横軸にプロットされているようです。こちらのデータで遊んでみます。

画面下の「Browse all Approved Drugs」から遷移した先のページで、「Small molecule」のみにフィルターをかけ、csv形式でデータをダウンロードしました。

Pandasで読み込んで中身を確認します。区切りがセミコロン(;)となっていたのでsepで指定して読み込みました。
import pandas ps pd df = pd.read_csv('CHEMBL25.csv', sep=";") df.shape # (1636, 32)
16363エントリー、32列の情報をもつテーブルとなりました。 欠損値も多くこのままでは使えなさそうなので、必要な列だけを残して絞り込もうと思います。
まず、「Drug Type」という列を確認してみました。
print(df["Drug Type"].unique()) # ['9:Inorganic' '1:Synthetic Small Molecule' '10:Polymer' '7:Natural Product-derived']
無機化合物やポリマーも含まれているようです。合成低分子に絞り込みます。
df_syn = df[df["Drug Type"] == "1:Synthetic Small Molecule"] print(len(df_syn)) # 1149
1149エントリー残りました。構造情報は「Smiles」列にあるようですが、まだNaNとなっている行があるので除きます。
df_syn_smi = df_syn.dropna(subset=['Smiles']) print(len(df_syn_smi)) # 1106
SMILESの情報をもつ合成低分子は1106エントリーでした。今回使用する情報として以下の4列を残しました。
① Parent molecule (化合物のChEMBL ID)
② First Approval (初めて承認された年?)
③ oral (経口投与か否か?)
④ SMILES
各列の情報の内容は私の勝手な推測です。このまま突き進みます・・・すみません。。。
df2 = df_syn_smi[["Parent Molecule", "First Approval","Oral", "Smiles"]] df2.head()
こんな感じになりました。

RDKitで構造の描画と指標の計算
とりあえずDataFrameができたので、RDKitを使ってSMILESをMolオブジェクトに変換し、分子量等の指標を求めます。 RDKitのPandasToolsの使い方は「化学の新しいカタチ」さんの記事を参考にさせていただきました。*2
from rdkit import rdBase, Chem, DataStructs from rdkit.Chem import AllChem, Draw, Descriptors, PandasTools from IPython.display import SVG # SMILESの列を指定して、Molオブジェクトの列を新しく追加 PandasTools.AddMoleculeColumnToFrame(df2, "Smiles") # Molオブジェクトから色々と指標を計算 df2['MW'] = df2.ROMol.map(Descriptors.MolWt) # 分子量 df2['LogP'] = df2.ROMol.map(Descriptors.MolLogP) # LogP df2['NHD'] = df2.ROMol.map(Descriptors.NumHDonors) # 水素結合ドナーの数 df2['NHA'] = df2.ROMol.map(Descriptors.NumHAcceptors) # 水素結合アクセプターの数 df2['NHetero'] = df2.ROMol.map(Descriptors.NumHeteroatoms) #ヘテロ原子の数 df2['NAR'] = df2.ROMol.map(Descriptors.NumAromaticRings) # 芳香環の数
こんな感じです。

注意点ですが、RDKitのPandasToolsは Pandas v0.25.1 以降のバージョンでは構造式の描画がうまくされず文字列になってしまうようです。イシュー (?)に書いてありました。*3
同様の症状がみられた場合は Pandas 0.25.0以前のバージョンにインストールしなおすと良いようです。私もこれで直りました。
最後に、時系列ならべるために「First Approval」でソートしました。
df3 = df2.sort_values('First Approval')
データの準備完了!
Bubbleチャートをプロット
本題のバブルチャートを作成します。
先の記事のパッケージ「bubbly」と「Plotly」のインストールが必要になります。
どちらも「pip install ~~」で大丈夫でした。
まずは必要なものをimport
import plotly from plotly.offline import init_notebook_mode, iplot init_notebook_mode() from bubbly.bubbly import bubbleplot
バブリー.バブリー・・・
以下の図では、各分子を
- X軸:分子量(MW)
- Y軸:LogP
- バブルのサイズ:芳香環の数(NAR)
とした設定でプロットし、継時的な変化を眺めてみます。さあコピペだ!
figure = bubbleplot(dataset=df3, x_column='MW', y_column='LogP', bubble_column='Parent Molecule', time_column='First Approval', size_column='NAR', x_range=[0,1000], y_range=[-5, 10], x_title='Molecular Weight', y_title='LogP', title='Changes of Drug Properties', scale_bubble=1) iplot(figure)
こんなんできました。
1980年代と90年代にポイントの数が一気に増える年があるようです。 また2010年代に入ってからバブルのサイズが大きくなったように見えました。芳香環の数が増えたのでしょうか?
もう少し情報を加えて、「Oralか否か」で色付けしてみたいと思います。 「True/False」のままではうまくいかなかったので、「1/0」に変換してプロットしました。
# 1をかけるとTrue/Falseが1/0に変換できるらしい df3['oral'] = df3['Oral']*1 # color_columnとcolorbar_titleの設定を付け足し figure = bubbleplot(dataset=df3, x_column='MW', y_column='LogP', bubble_column='Parent Molecule', time_column='First Approval', size_column='NAR', color_column='oral', x_range=[0,1000], y_range=[-5, 10], x_title='Molecular Weight', y_title='LogP', title='Changes of Drug Properties', colorbar_title='Oral', scale_bubble=1) iplot(figure)
色付けは基本的に「青色(0; oral = False)」「黄色(1; oral = True)」となっています。
が、どうも不具合があるようで、その年にカテゴリの一方しかない場合は青色・黄色以外の色となっています。
また、最初の再生ではカラーバーもカテゴリーの数に合わせて変化していますが、一度Pauseするとカラーバーがその時点で固定化されてしまいプロットにおける色を正確に反映しなくなってしまいます。
ですので色は「大体そんな感じ」くらいでとらえてください。(私の能力では修正できません。すみません・・・)
なんとなく、2000年代後半から黄色が増えたように思います。
抗体医薬等が承認されるにつれて「低分子は経口投与を重視」というようになったのでしょうか?
雰囲気だけで適当なことを言っています。フェイクニュースです。
化合物空間でプロット
さて、ここまできたらついでに化合物空間における変遷までみてまいましょう。
毎度コピペ記事で申し訳ありませんが@iwatobipen先生のAdvent Calendar記事 からコードを拝借し、フィンガープリントをt-SNEで2次元に落としてプロットします。*4
まずは時系列関係なくエントリー全てをつかって化合物空間を作ります。
import matplotlib.pyplot as plt import seaborn as sns from sklearn.manifold import TSNE # フィンガープリントを格納するための空のリスト fps = [] # DataFrameのMolオブジェクトを取り出して順番にフィンガープリントを計算しリストに追加 mols = df3['ROMol'] for mol in mols: fp = AllChem.GetMorganFingerprintAsBitVect(mol, 2) arr = np.zeros((1,)) DataStructs.ConvertToNumpyArray(fp, arr) fps.append(arr) # numpyのarrayに変換 fps = np.array(fps) # t-SNEで次元圧縮 tsne = TSNE(random_state=123) res = tsne.fit_transform(fps) # プロット plt.clf() plt.figure(figsize=(12, 6)) plt.scatter(res[:,0], res[:,1])
こんな感じで散らばりました。

この空間が時代とともにどのように埋められていったのでしょうか??
DataFrameにt-SNEでえられた2次元プロットのXとYの値を追加します。
df3['tSNE_X'] = res[:,0] df3['tSNE_Y'] = res[:,1]
X、Y軸はtSNEのプロットそのままに、バブルのサイズを「分子量(MW)」、色は「oral(True/False)」としてバブルチャートを作成してみます。
figure = bubbleplot(dataset=df3, x_column='tSNE_X', y_column='tSNE_Y', bubble_column='Parent Molecule', time_column='First Approval', size_column='MW', color_column='oral', x_range=[-60,60], y_range=[-60, 60], title='Changes in the Chemical Space', colorbar_title='Oral', scale_bubble=1)
あいかわらず色のバグはそのままで申し訳ありませんがこんな感じになりました。
予想以上に満遍なく全体にちらばりました。 時系列に従ってポイントの位置が集まる場所が見えたりしたら面白いかな?と思ったのですが、そんなに綺麗にはいかないですよね・・・
おわり
以上、バブルチャートをつかって合成化合物の変遷を眺めてみました。 これといった中身がなく申し訳ありませんが、なんだかバブリーだからオッケー!!
・・・というのもあれなので真面目な動画のページもご紹介させていただきます。
Norvartis社 Mike Shultz 氏によるウェビナー動画で、Hulinks社のページにありました。
URLはこちら です。StarDropというソフトウェアの使用例の紹介のようです。*5
動画の中で「Drug-likeness」について、過去20年の承認薬の解析をもとに「Rule of 5」をいかに更新していくか、といった議論がされています。
経口薬の分子量は増大の傾向にあるものの、cLogPと水素結合のドナーの数(HBD)は変化していないそうな・・・
(日本語字幕がなくてよくわからなかったので雰囲気です。すみません。)
・・・なるほど
・・・おしまい!!
色々と間違いがあると思います。ご指摘いただければ幸いです。
低分子によるオリゴマーの制御? 〜低分子は神経変性を抑制できるか?〜
創薬 Advent Calendar が空いていたので埋めます。
今年は @fmkz__先生と@iwatobipen先生によるpy4chemoinfomaticsに始まり、 坊農先生の生命科学者のためのデータ解析実践道場、 金子先生の化学のための Pythonによるデータ解析・機械学習入門 の出版と、in silico創薬に興味があっても具体的にどうすれば良いか分からない勢にとってはとてもありがたい一年でした。 インターネットで検索すれば色々な情報が手に入りますが、書籍という一貫した形で情報を得ることができるというのはやっぱり密度が異なるように思います。(私は旧世代なので紙でしか長文を読めないです・・・)
Dryの書籍の目玉は上にあげたものとして、Wetの書籍での今年の目玉はやはり白木先生の相分離生物学だったのではないでしょうか? 分子レベルと細胞レベルの階層を繋ぐ新たな視点、相分離について日本語での系統だった解説書ということで新しい分野の始まりの興奮を感じる大変素晴らしい書籍でした。
「相分離メガネ」を手にしたところで、ちょっと変わった(?)創薬のアプローチ「低分子によるオリゴマーの制御」をご紹介したいと思います。
- 注) 以下の記事では臨床試験段階の医薬品をご紹介します。実際に効果があるのかまだ検証段階であり、全ての情報が公開されているわけではないため私の推測を含むものとなっております。また、ご紹介する内容は私とは一切関係のない企業ということをご了承ください。
- Parkinson病って?
- Parkinson病とα-synuclein
- Parkinson病と2つの仮説
- αシヌクレインのチャネル形成による毒性発現モデル
- Neuropore Therapiesにおける低分子開発のアプローチ
- まとめ
今回ご紹介するのは NEUROPORE THERAPIES Inc.が見出したParkinson病を標的疾患とする低分子 UCB0599で、 2019年現在UCBにより米国で臨床開発(Phase 1B)が実施されています。
さて、こちらの低分子、Parkinson病において関連性を指摘され名高いタンパク質 α-synuclein のオリゴマー化を妨げることがその作用機序(MOA)としてうたわれています。 どういうことなのか?Nuropore Therapies社による文献をもとに彼らのアプローチをたどってみたいと思います。
- A de novo compound targeting α-synuclein improves deficits in models of Parkinson’s disease Wrasidlo, W. et. al. Brain 2016(139)3217
- The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson's disease. Price, DL. et. al. Sci. Rep. 2018(8)16165
** どちらもオープンアクセスとなっています。
Parkinson病って?
そもそもParkinson病って?ということを少しだけ・・・難病情報センターのページより引用させていただきます。
パーキンソン病 1.「パーキンソン病」とはどのような病気ですか
振戦(ふるえ)、動作緩慢、筋強剛(筋固縮)、姿勢保持障害(転びやすいこと)を主な運動症状とする病気で、50歳以上で起こる病気です。時々は40歳以下で起こる方もあり、若年性パーキンソン病と呼んでいます。 (中略)
4.この病気の原因はわかっているのですか
大脳の下にある中脳の黒質ドパミン神経細胞が減少して起こります。ドパミン神経が減ると体が動きにくくなり、ふるえが起こりやすくなります。ドパミン神経細胞が減少する理由はわかっていませんが、現在はドパミン神経細胞の中にαシヌクレインというタンパク質が凝集して蓄積し、ドパミン神経細胞が減少すると考えられています。このαシヌクレインが増えないようにすることが、治療薬開発の大きな目標となっています。
文字で読んでも良くわからないですね。症状を動画で見ていただくのが一番と思います。
What do the symptoms of Parkinson's look like?
主たる特徴としては運動障害があげられ、動画をご覧いただいてもお分りいただけると思いますが通常であれば問題なく行える、物をつかむ、歩く、といった動作が行えなくなります。高齢者で多くなる疾患であることからこれまでできていた日常の動作が(徐々に)うまく行えなくなるという症状は肉体だけでなく精神的にも大変苦痛を強いると想像できます。
アルツハイマー病とならぶ神経変性疾患としてあげられることも多いパーキンソン病ですが、現在の主な治療方法は対症療法(症状を緩和させる治療方法)で疾患そのものを治療することは依然として困難です。ドパミン神経が減少することから、神経伝達物質であるドパミンの補充を目的とし、ドパミンの作用する受容体であるドパミン受容体に対するアゴニスト、あるいはドパミン前駆体であるL-ドパを投与するというのが薬理学の教科書でとりあげられる主な内容でしょうか?
残念ながら治療の現場の実際を私は把握していないためうろ覚えの知識ですみません・・・
Parkinson病とα-synuclein
さて引用のように、Pakinson病については患者さんのドパミン神経細胞にレビー小体と呼ばれる特徴的な構造体が見られること、その主な構成要素がαシヌクレインであることから、αシヌクレインが疾患の主たる要因ではないかということが示唆されています。 タンパク質の異質化、蓄積による神経変性疾患として、アルツハイマー病におけるアミロイドβと同様取り上げられることの多いαシヌクレインですが、ではこのタンパク質が「生体内、細胞で実際にどのような機能を果たしているのか??」というと未だ良くわかっていない点が多いようです。
現段階で示唆されているのはαシヌクレインが複数集まり凝集化(オリゴマー化、さらに高次の凝集体形成)すると毒性が生じ、細胞死を起こすのではないかということのようです。じゃあなんで凝集体を形成するの?ということに関しては未だ不明で、酸化ストレスによるタンパク質の変性などが議論されています。
余談ですがParkinson病で欠落するドパミン神経の生産するドパミンの構造、有機化学を齧った身からするととても不安になる構造です。

ベンゼン環にヒドロキシ基がオルト位でジ置換・・・如何にも酸化に弱そうなアラート構造そのものです。酸化によるキノン構造の形成、タンパク質との共有結合による変性、毒性の発現といった推測をしてしまいます・・・。
Parkinson病と2つの仮説
さてParkinson病とαシヌクレインの関係性は示唆されて久しい訳ですが、ではなぜこの疾患が進行性なのか?凝集体が形成されるにしてもなんでそれが伝搬していくの?ということに関しては大変興味深い2つの仮説が提唱されています。 まずはParkinson病はプリオン病の一種ではないかという仮説です。こちらについては2016年のNatureの記事をご覧いただくのが分りやすいか思います。*1
Pathology:The prion principle Nature2016(538)S13-S16
上記記事の図がわかりやすので是非参照していただきたいのですが、αシヌクレインのモノマーがミスフォールドし「オリゴマー化 → 繊維化 → 凝集」することにより細胞毒性を発現すること、 ミスフォールドしたタンパク質が「クロイツフェルト・ヤコブ病(狂牛病)」のようにプリオンとして働き、次々に同様のミスフォールドと凝集を誘導することで疾患が進行するのではないか、 という仮説が議論されています。
同様の神経変性疾患における「プリオン病仮説」はアルツハイマー病においても示唆されています。アルツハイマー病の治療における最近の大きな話題としてはエーザイとバイオジェンによるアデュカブマブのFDAへの承認申請( プレスリリース)があげられると思いますが、私はアルツハイマー病に対する抗体医薬というアプローチを知った際にとても驚きました。
中枢疾患の代表とも言えるアルツハイマー病に対して抗体医薬!!!どういうこっちゃ????抗体って中枢に行かないんじゃないの??そもそも細胞内のタンパク質を抗体で標的って無理じゃないの???となっていしまいました。 ですが、上記プリオン仮説で変性タンパク質の伝搬による疾患の進行という仮説を知り、なるほどそれで血中の変性タンパク質を抗体でねらうという発想になるのかーと腑に落ちました。(残念ながら私は臨床試験の治療効果の確からしさを判断するほどの知識がありません・・・)
では、Parkinson病の進行が変性したαシヌクレインの伝搬によるものとして、その根拠となるような情報はあるの?ということになると思いますが、この点に関してBraakの仮説というものが提唱されています。*2
2003年にHeiko Braakにより提唱されたParkinson病の病態の進行に関する仮説で、末梢における変性αシヌクレインが嗅球、迷走神経から脳幹、大脳へと徐々に伝搬することで病変が広がっていくのではないかとする仮説です。 Pakinson病において特徴的な運動障害に先行・並行して非運動症状(自律神経障害、睡眠障害、レム期睡眠行動異常症(RBD)、認知機能症状)の進行が見られることからも幅広く議論されている仮説です。*3
未だ仮説の段階ですが、中枢における神経変性疾患において原因とされるタンパク質の変性と末梢における病変の関連性については一定の議論がなされているということはお分りいただけたと思います。
以上を踏まえた上で本題、「低分子によるタンパク質オリゴマーの制御」についてご紹介したいと思います。
αシヌクレインのチャネル形成による毒性発現モデル
αシヌクレインがParkinon病との関連していることは一定のコンセンサスが得られているとして、では一体どんなメカニズムで細胞毒性を発現しているの??というのが次の疑問になると思います。 少々古い文献で恐縮ですが一つの仮説として2010年Fantaniらによるレビュー*4で、αシヌクレインがオリゴマー化することで細胞膜上にチャネルを形成、カルシウムイオンの細胞膜透過性を異常化させることで恒常性の破綻、細胞死を誘導するというモデルが紹介されています。
今回ご紹介するNeuropore Therapiesによるアプローチは上記のαシヌクレインの細胞膜上におけるオリゴマー形成による毒性発現を低分子により阻害することで治療を目指すというものになります。 現在臨床開発中であり実際に治療効果があるのか、という点については検証中の段階ですが既存のアプローチが対症療法しか存在しないパーキンソン病において疾患の進行を抑制する可能性を含んだ一つのアプローチとして大変興味深いアプローチです。
Neuropore Therapiesにおける低分子開発のアプローチ
では文献の中身を見てみましょう。Brain 2016(139)3217 では初期化合物 NPT100-18Aの創製にいたった経緯が紹介されています。
まず、著者らはαシヌクレインのオリゴマー化に関連する部位としてαシヌクレインのC末端ドメインに着目し、96-102残基を低分子により模倣するというアプローチで化合物をデザインしています。 96-102残基が別のαシヌクレイン 80-90残基と相互作用することで2量化(ダイマー形成)することがオリゴマー形成の出発点との仮説をもとに、 配列 KKDQLGK をもとにしたモデリングから環状アナログをデザイン・合成、NPT100-18Aを見出しています。
詳細については上記文献の Supplementary fig 1をご参照ください。また、化合物の変遷に関しては上記文献 Supplementary fig 2に掲載されています。
モデリングと組み合わせたタンパク質残基の模倣による低分子デザインというだけでも非常に興味深いアプローチですが、化合物の評価方法としても大変興味深いアプローチが取られています。
通常、新しい化合物の評価方法としては標的のタンパク質の機能に基づいた評価が行われると思います。例えばGPCRであればシグナル下流のcAMPの変動、キナーゼであれば標的タンパク質のリン酸化の変動といったところになるでしょうか? では同一タンパク質のオリゴマー化、凝集に対する抑制効果を評価するにはどうすれば良いか?著者らは一つのアプローチとしてNMRシグナルの変化を用いています。
詳しい内容は上記文献 Figure 2 を参照していただきたいのですが、細胞膜を模倣したリポソームとαシヌクレインを混合した系において化合物を添加し、 化合物がオリゴマー化を阻害すればαシヌクレインが膜上に留まらず単体として溶液に遊離した状態が保たれるとの仮説のもと、αシヌクレイン残基のNMRシフトの変異を観測しています。(Brain 2016(139)3217 fig 2)
上記評価にもとづき、リポソーム未添加の単量体αシヌクレイン、リポソーム添加におけるαシヌクレイン、リポソームと化合物 NPT100-18Aを共に添加したαシヌクレインのNMRシフトを比較、 化合物添加によりNMRシフトがαシヌクレイン単独の際に戻ることを確認し、その効果を評価しています。なるほどこんなin vitro評価方法があるのか!という非常に興味深い方法となっています。
より高次な評価としては膜上での染色を利用したリング構造(チャネル形成)の形成を阻害する効果が議論されています。( Supplementary fig 3)
非常に興味深いアプローチでオリゴマー化を阻害する低分子 NPT100-18Aを見出したNeuropre Therapies社ですが、臨床に進むには課題(半減期、中枢移行性など)があったようで、 さらなる検討の結果 NPT-200-11を見出しin vivo マウスモデルにおける治療効果を報告しています*5。
文献中にはNPT-200-11の具体的構造は開示されておらず、またこちらが臨床開発中のUCB0599そのものかかはわかりませんが、 NPT100-18A同様にペプチドの一部を環化させることで膜透過、血中安定性といった課題をクリアしているとすれば非常に興味深い低分子デザインのアプローチといえるのではないでしょうか?
まとめ
以上、大変粗い紹介で恐縮ですが神経変性疾患における原因療法を目指した一つのアプローチ、「低分子によるオリゴマ−化の制御」でした。 くり返しですが、未だ臨床開発初期の段階で疾患の特徴(長期にわたりゆっくりと進行する疾患であり、早期における介入が治療効果の発揮が重要と想定されるが、診断・治療効果の判定も含めて早期介入、効果の実証が難しい)を含めて、 臨床で使用される可能性についてはまだまだ未知数のアプローチですが大変興味深いアプローチとして紹介させていただきました。
無理やりですが、標的タンパク質1分子ではなく、それらが複数集まったオリゴマー・凝集体を制御するアプローチは生体内におけるタンパク質相互作用に基づく異質化、相分離の制御にもつながる新たなアプローチとして興味深いのではないのでしょうか?
高齢化に伴い患者数の増加が予想されるParkinson病については「創薬」というカテゴリーに収まらず、患者さんの生活の改善にむけたアプローチが検討されています。 例えば次の記事では、Parkinson病患者における歩行障害(歩き始め、一歩を踏み出すのが困難)を解決する手段として、 靴にレーザー発光機能をとりつけることで歩行を補助するというアプローチが紹介されています。
This laser helps Parkinson's patients walk again
またLIFTWAREの開発するこちらのデバイスは運動障害(手の震え)に悩む患者さんの食事を補助するため、振戦に関わらず使用できる食器(スマートデバイス)が実用化されています。
これらのアプローチを知ったとき非常に感銘を受けました。LIFTWAREの掲げる「Eat with confidence」は大変素晴らしものと思います。 徐々に運動機能が奪れていくという、これまで自分でできていたことができなくなるという疾患のもたらす苦痛は大変大きなものと推察されています。
生活の根源である衣食住が自分自身でできること、健康な人間にとっては当たり前と思えることができなくなること、それは自分自身が自分自身で無くなるような苦痛を強いるものと思います。あたらしい創薬アプローチに限らずテクノロジーの進化によって患者さんの日常生活が少しでもより良いもの、自信がもてるようになるというのはとても重要なことだと思います。
アルツハイマー病・パーキンソン病を含めた神経変性疾患はいくつかの対症療法は存在するものの、今なお有効な原因療法はなく細胞療法も含めた新規なアプローチが検証されている段階です。まだまだ時間はかかるでしょうが少しづつでも患者さんの過ごしやすい未来にむけて科学が進歩していくことを期待しています。
中途半端な知識で書いてしまいました。誤り等ありましたらご指摘いただければ幸いです。
※ Creative Commonsかわからない文献の図を引用してしまっていたのでFigureを消しました。本文中の文献はすべてアクセス可能なものでしたので適宜ご参照いただければと思います。
*1:Pathology:The prion principle Nature2016(538)S13-S16
*3:「専門医が知っておくべきParkinson病の病態と治療の展望」神経治療 34:182-187 2017
*4:Expert Reviews in Molecular Medicine 2010(12)e27
*5:Sci. Rep. 2018(8)16165
HomebrewとAnacondaとRDKitを入れ直した話
3連休初日ですが皆さまいかがお過ごしでしょうか?私は連休続きで週5日勤務に耐えられない身体になってしまいました・・・
さて、CBIにもケモインフォ討論会にもいけなくて残念に思っていたところ、私のようなパソコン苦手な初心者にとってはとても嬉しい書籍が2冊出版されました!
坊農先生の「生命科学者のためのデータ解析実践道場」

- 作者:坊農秀雅
- 発売日: 2019/09/27
- メディア: 単行本
金子先生の「化学のためのPythonによるデータ解析・機械学習入門」

- 作者:弘昌, 金子
- 発売日: 2019/10/23
- メディア: 単行本(ソフトカバー)
買うしかない!ということで早速、amazonでポチッとしたあと、久しぶりにターミナルで 「conda install XXXX」 したらMacが異音を発生してターミナルがおかしくなりました・・・
brewもcondaも「comand not found・・・」
・・・で、泣きながらもう一回環境構築(?)したので、備忘録を残します。
やったこと・・・
- HomebrewとAnacondaをインストール
- RDKitのインストール
- Jupyter notebookからRDKitを使えるようにする
- pymolのインストール
- (おまけ)古いHomebrewとAnacondaの削除
- おしまい
私のパソコン:「MacBook Pro(2017) / macOS High Sierra バージョン 10.13.6」
HomebrewとAnacondaをインストール
こちらの記事をそのままやりました。(引用記事ですみません)*1
# Homebrewのインストール /usr/bin/ruby -e "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/master/install)" # Hombrew のアップデート brew update --force && brew upgrade # pyenvのインストール brew install pyenv echo 'eval "$(pyenv init -)"' >> ~/.bash_profile exec $SHELL -l
このあと「.bash_profile」をテキストエディタで開いて追加で書き込みをして以下のようにしました。
export PYENV_ROOT="$HOME/.pyenv" export PATH="$PYENV_ROOT/bin:$PATH" eval "$(pyenv init -)"
「export ~~~」の2行を書き込まないとうまく動かなかった・・・
ついで、Anacondaをインストール。 RDKitは「Python 2系はもうサポートしないよ!」、ということなのでPython3系で新しそうなバージョンを入れました。
# インストール可能なバージョンの確認 pyenv install -l # Python version 3系のをインストール pyenv install anaconda3-5.3.1 pyenv global anaconda3-5.3.1
インストールできました!が、バージョンが新しいAnaconda (4.4以降?)はこのままでは使えないらしく、 ターミナルで「configureをちゃんとしなさい!」みたいなエラーが出ました。
エラーメッセージにしたがって、echoを実施して「.bash_profile」を修正したら使えるようになりました。
# 現在のユーザーがcondaを使えるようにする。 # 下の行の「XXXXX」は実際にはユーザーの名前が入っています。アホすぎる名前で恥ずかしいので伏せてます。 echo ". /Users/XXXXX/.pyenv/versions/anaconda3-5.3.1/etc/profile.d/conda.sh" >> ~/.bash_profile # condaのbase (root) environmentをPATHにずっと追加するようにする echo "conda activate" >> ~/.bash_profile
ここまでやって、「.bash_profile」は以下の状態になっていました。
export PYENV_ROOT="$HOME/.pyenv" export PATH="$PYENV_ROOT/bin:$PATH" eval "$(pyenv init -)" . /Users/XXXXX/.pyenv/versions/anaconda3-5.3.1/etc/profile.d/conda.sh conda activate
「/conda.sh」の行が書いてあれば「"$PYENV_ROOT/bin:」の行は不要?みたいな記述も見かけましたが とりあえず動いているのでそのままにしてます。 ・・・私みたいな初心者にはPATHとか依存とか意味不明でつらいです。
以上でHomebrewとAnacondaの準備終了です。
RDKitのインストール
RDKitのドキュメントと 「化学の新しいカタチ」さんの記事にしたがってRDKitをインストールします。*2*3
# Pythonのバージョンを確認 python --version # pythonのバージョンを指定して RDKitをインストール conda create -c rdkit -n my-rdkit-env python=3.7 rdkit # 新しく作ったconda 環境(my-rdkit-env)をアクティベート conda activate my-rdkit-env
これでRDKitもインストールおしまい。
ですが、初心者の味方 Jupyter notebook からRDKitを利用するためにはもう一手間が必要です。
Jupyter notebookからRDKitを使えるようにする
ここまでの設定のまま Jupyter notebook から RDKit を import しようとしても「ModuleNotFoundError」というエラーが出ます。
どうもこちらの記事 によると、RDKit用につくった仮想環境(my-rdkit-env)にはjupyterをまだインストールしていないので、デフォルトのbase環境(?)のjupyter が起動されていることが原因のようでした。*4
対処方法としては2通りありそうでした。
方法①: 新しい仮想環境をactivateしたあとで、jupyterをインストールする
方法②: jupyter notebook上でカーネルを切り替えられるようにパッケージをインストールする。
今回は方法①を試した後で、方法②のパッケージもインストールしてみました。
# RDKitの仮想環境がactivateされている状態でjupyterをインストール conda install jupyter
この後、jupyter notebookを起動したら無事RDKitをimportできました!
ついでにこちらの記事 を参考にカーネルを切り替えるパッケージをインストールします。*5 引用記事そのままで恐縮ですが・・・
# パッケージのインストール pip install environment_kernels #jupyterの設定ファイル作成 jupyter notebook --generate-config
上の「environment_kernels」のインストールの際に「PyHamcrestが入ってないよ!」みたいに怒られた時は「pip install PyHamcrest」を先にやってから、もう一度インストールすれば良いみたいです。
で、作成した設定ファイル「~/.jupyter/jupyter_notebook_config.py」をテキストエディタで開いて最後の行に以下を追記・・・
c.NotebookApp.kernel_spec_manager_class = 'environment_kernels.EnvironmentKernelSpecManager' c.EnvironmentKernelSpecManager.conda_env_dirs=['. /Users/XXXXX/.pyenv/versions/anaconda3-5.3.1/envs/']
この後で記事に従って念のため
pip install jupyter
とやったら、「もう入ってるよ!」と言われました。・・・ですよね。
jupyter notebookを起動して確認してみます。

新しいNotebookを作る際にどの環境を使えるか選べるようになりました!試しに「Environment(conda_my-rdkit-env)」を選択して新しいnotebookをつくりrdkitをimportしてみます。

警告出てますがとりあえずこれでも無事動きました!ついでにもう一つの仮想環境「Environment(conda_anaconda3_5.3.1)」では・・・

となりました。ようやくcondaの仮想環境という考え方がちょっとだけわかった気がします。
pymolのインストール
RDKitがインストールできたので、ついでにPymolもインストールしておきます。
brew tap brewsci/bio brew install pymol
おしまい。
(おまけ)古いHomebrewとAnacondaの削除
ちなみに古い環境の削除ですが、PATH(?)がおかしくなっているのかターミナルを起動したホームディレクトリからはHomebrewのアンインストールコマンドが使えませんでした。
どうもHomebrewは「Cellar」という名前のフォルダに入っているようなので、ディレクトリを移動してからコマンドをコピペしたらアンインストールできました。*7
# それっぽい場所に行く cd /usr/local/Cellar # アンインストールコマンドをコピペ ruby -e "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/master/uninstall)"
ここに行き着くまでに2週間かかった・・・・。
Anacondaは「anaconda-clean」というので削除するのが正当法(?)のようでしたが、 どうにもうまくいかなかったのでそれっぽいフォルダをゴミ箱に放り込んで捨てました。*8
おしまい
以上、環境構築の備忘録でした。何番煎じか分からないコピペ記事で申し訳ないですが備忘録ですのでご容赦ください。
なんとか動く感じになったので先生がたの本を読み始めたいと思います。
記載の誤り等ありましたらご教示いただければ幸いです。
っていうかRDKitの jupyter extensionとは一体?調べても分からなかった・・・
*1:https://qiita.com/aical/items/2d066801a7464a676994
*2:http://www.rdkit.org/docs/Install.html#cross-platform-under-anaconda-python-fastest-install
*3:https://future-chem.com/rdkit-intro/
*4:https://weblabo.oscasierra.net/python-anaconda-jupyter-modulenotfounderror/
*5:https://qiita.com/yuto_ohno/items/4f5b58978235c35fbe7e
*6:https://qiita.com/Ag_smith/items/58e917710c4eddab46ee
*7:https://qiita.com/uhooi/items/91d2261ab30b6f2c143a
*8:https://weblabo.oscasierra.net/python-anaconda-uninstall-macos/
MITのオンラインコースでお勉強した話
随分とお久しぶりです。 6月から9月にかけて約3ヶ月間、オンラインコースで機械学習のお勉強をしてみました。 折角なので内容をご紹介したいと思いいます。

- MIT MicroMasters Program
- MicroMasters program in Statistics and Data Science
- なぜ受けることにしたのか?
- 内容
- 感想
- もっと感想
- まとめ
MIT MicroMasters Program
MicroMasters program はMITが edX というオンラインの学習プラットフォーム上で提供しているプログラムです。 MITのページ によると 「データサイエンティストの需要の拡大に応える」ために「働きながらでも統計学とデータを厳密な方法で勉強できるように機会を提供する」 と言うのが目的のようです。
修士課程の小さい版(?)ということで、分野と内容を絞ってコースが提供されています。 コースにもよりますが、基本的に無料で受講することが可能で、受講期間内であれば講義のビデオを繰り返し見たり、 オンライン上で問題を解いたりすることができます。但し、修了証明書を手に入れたり一部テストを受けるには授業料が必要になります。
MicroMasters program in Statistics and Data Science
MicroMasters program in Statistics and Data Scienceは 5つのコースからなっています。
- Probability - The Science of Uncertainty and Data
- Data Analysis in Social Science - Assessing Your Knowledge
- Fundamentals of Statistics
- Machine Learning with Python: from Linear Models to Deep Learning
- Capstone Exam in Statistics and Data Science
コース 5. は 1. - 4.のコースを受けた後の総括のテストのようなものなので、実質的に受ける授業としては4コースでしょうか?
全てのコースが一年中受けられる訳ではなく、各コースの開始 - 終了の日程が決められています。
さらに、修了証を手に入れるためには期間の中に設定された締め切りまでに、授業と宿題を進めていく必要があります。
(「Lecture 5 / Homework 3の deadlineは 07月20日だよ〜〜」みたいなのが設定されています。「君ら提出せんから延長するわ〜〜」みたいな感じで、割と2,3日期間は延長されます。)
気になるお値段ですが、1コース $300 USDです。5コース受けるとセット割引的なものもあるようですが中々のお値段・・・
今回、私は6月開講の 4. 「Machine Learning with Python: from Linear Models to Deep Learning」を受講しました。 絶対に途中で放棄する自信があったので、逃げ道を断つべく払いましたよ!300ドル! やる気はお金で買うんだ!(ダメ人間)
なぜ受けることにしたのか?
実はオンラインコースに挑戦するのは初めてではありません。 以前にMOOC (Massive Open Online Courses) の大手、CouseraでAndrew Ng先生の「Machine Learning」を受けようとしたことがあります。 ですが、優しい語り口の誘う眠気と、英語のハードル、Octave / Matlab (?)を覚えないといけない、ということでイントロで撃沈してしまいました・・・。
性懲りもなくまたオンラインコースに挑戦したのは、全くもって偶然に再開した旧い知人にこのコースを紹介されたからです。 この知人というのがとんでもなく優秀かつ出来る男なので、同じコースを受けたら私も賢くなるのでは!と思ってしまったんです。(・・・浅はか)
いくつかあるコースから「Machine Learning with Python: from Linear Models to Deep Learning」を選択したのは、 「Python」&「機械学習」という流行りに乗っかっとくか、という理由です。(・・・やっぱり浅はか)
内容
さて、肝心の内容です。カリキュラムは以下となっていました。
- Unit 1 「Linear Classifiers and Generalizations」 (2 weeks)
- Unit 2 「Nonlinear Classifications, Linear regression, Collaborative Filtering」 (2 weeks)
- Unit 3 「Neural networks」(2.5 weeks)
- Midterm Exam (1 weeks) ← 課金した人だけ
- Unit 4 「Unsupervised Learning」 (1 week)
- Unit 5 「Reinforcement Learning」(2 week)
- Final Exam (1 weeks) ← 課金した人だけ
各ユニットはさらに複数のLecture と Homework、Projectから構成されています。

Lecture
Lectureは 各 5 ~ 20 分くらいの動画と付随するクイズ(1-2問)からなるセット、5〜6個から構成されています。 (全部でLecture 19個)
一つのページに、動画とクイズがあり、さらにDiscussion Forumと呼ばれる掲示板のようなものがあります。 ここでは、受講生が互いに議論したり、TAが質問に答えてくれたりしてくれます。 クイズやテストの回答そのものを書き込むことは禁止されていますが、 問題で躓くポイントや、解決手段、参考になるリンクを共有してくださる方がいらっしゃったので、 私はひたすらこの掲示板をカンニングしながらクイズと宿題に取り組んでいました。
「3回まで間違えてもOK!」とかそういう感じで設定されていて、なのに「選択肢:2つから選べ」というクイズだったりして、設計ミスの疑惑もありましたが、 クイズは理解を促すという目的なので優しい設定だったのでしょう。
Homework
Homework はいくつかの Lecture の内容をまとめた問題で、先のクイズよりも難しくなっています。 (全部でHomework 7個)
大雑把なイメージですが、Lecture の動画で理論やアルゴリズムの説明があり、Homeworkは具体的な数字やグラフを当てはめて実際に解いて見ましょう、という感じです。
Project
Project は Lecture、Homework の内容を踏まえてより複雑な問題設定で取り組んで見ましょうという形式です。 (全部でProject 5個)
Pythonを使ってプログラミングを行うのは主にこのProjectでした。
具体例で行くと、「Collaborative Filtering」ではお題として「映画をオススメするシステムを作りましょう」といった大枠が設定されます。(Netflix関連??)
これだけだとどこから手をつけて良いかさっぱりわからん・・・となってしまいますが、ご安心ください。
データセットとプログラム全体の枠組みは予め用意されていて、その中で使われている関数の中身を指示に従ってstep by step で実装していけば出来上がるように作られています。
つまり、受講生は果たすべき機能と引数のみ設定された一つ一つの関数の肉付け、「アルゴリズムをどうPythonで書き下せば良いか?」に集中すればOKとなっています。
(・・・とはいえこれが私にとってはメチャクチャしんどかったです。)
MicroMastersのコース紹介には「大体、週10 - 14 時間」取り組むことになりますよ。」と、書かれていましたが、 私の場合は平日ほとんど機能停止しているため、土日両日丸々つぶしてやっと、という感じでした。(体感では20時間くらい?)
感想
ごまかしごまかしなんとか証明書を獲得したので、偉そうに感想を書きます。・・・最低レベルの体験談として聞き流してください。

アルゴリズム?
個人的にもっとも良かった点は、パラメータを順次更新していくことで最適化するという、アルゴリズムで問題を解くというプロセスの理論と具体例を繰り返し実践できたことです。
私は数学が大の苦手で、高校数学でとまってしまっている(既にそれも危うい)のですが、高校までの数学で問題を解くといった場合、 数式をなにやら式変形して特定の解を導くといったことがほとんどだったように思います。 また、大学進学後も微分・積分、線形代数といった授業はありましたが、方法としては似たようものでした。(薬学部だったからかもしれません)
なので、今回edXで学んだような、更新のルールとそれが収束する理由が説明されて、 あとは繰り返しループを回して実際に逐次値が安定化して行くという経験をしたことがありませんでした。 ひょっとすると大学で、工学部等に進学されて最適化数学(?)を学ばれたのではない方は私と同じような状態なのではないかと思います。
「本当にこれで収束するのか!」「確かにループを回す解き方ならプログラミングでコンピューターにやらせるしかないな!」というのを納得できたのは非常に良い経験でした。
数学力?
また、Pythonでプログラミングを行おうとして、数学力がいるというのを実感しました。
行列同士の処理を行おうとした際に、「要素同士を掛け算して〜、足し合わせて〜」という処理を行いたいとして、
「これはなんか良い感じで内積とれば良い感じになるのでは??」とは思うのですが、結局どうして良いかわからず、
for ループを回してインクリメントしつづけるということを繰り返していました。
模範解答と比較した、自分のfor ループを回し続ける方法の遅さたるや!! Macが雄叫びをあげておりました・・・
ビデオ講義 ?
オンラインで講義を受けることのメリット・デメリットですが、スライド講義の場合は少しわかりづらいと感じました。 「Here, xxx *** ~~~~」みたいな感じで説明されるのですが、Hereってどこ!今どこの話しているの??となりました。 私はアナログ人間なので黒板に板書をしながら説明されているところを撮影した動画の方が流れを追いやすかったです。
一方で、繰り返し動画を見ることができる。高次元のグラフが綺麗に描写され、収束の様子がムービーで提示される。 何より、自分で自宅から授業を受ける時間を選ぶことができるという点は代え難いメリットだと思いました。 とりわけ地方在住のへっぽこ会社員にも新しい分野を勉強する機会が手軽に手に入るというのは大きいです。
もっと感想
で、これだけで「機械学習 & Deep Learning & Pythonすべてばっちり!」かというとそういうわけには行かないかな、という感想です。機械学習の話題だけでいえば「はじめてのパターン認識」の方が広く取り扱っているようにも思いました。実際に問題を解く上で、Lectureで解説されている内容だけでは足りず、「はじパタ」等を参照たりググったりしてやっと意味がわかるということがありました。
(Discussion ForumでもLecture と Homework・Projectの内容があっていないとの意見がありました)
ですが、私の場合は「はじパタ」単独では難しすぎて理解できなかったので、Lecture形式で順をおって説明されたことでやっと内容がわかったことがたくさんありました。 (例えば、k-MeansとK-medoidsの違いとか。。。) Lecture形式の良い点は躓きやすい点に力点を置いて説明してくれることだと思います。やっぱり教科書で字面を追うのとは違います。
Deep Learningについて、私は全く詳しくないのですが多分同じ感じなのかな?と思います。話題の網羅性だけなら、専用のコースや書籍を読む方が良いのでしょう。
また、Project等で取り扱われた問題も、既存のライブラリを使えば簡単に解けるものだと思います。
一方で、このコースでは、同じ問題 (例えばMNIST) を単純な線形判別・パーセプトロンからはじめてDLへと一貫して追って行くことができ、また、アルゴリズムを自分でプログラムに書く経験ができる点で理解が深まり良いと思いました。
私は教科書を読んでみても、実際に使うところまで落とし込むことはできないので、締め切りに追われて強制的に手を動かしてやっと少しわかりました。すみません・・・
まとめ
以上、オンラインコース受講のとりとめのない感想でした。
総じて質の高いコースだったので、興味を持たれた方は無料の講義をいくつか眺めてみられてはいかがでしょうか?課金するかどうかを決める期限は、開講後一ヶ月後程度に設定されているので、とりあえず眺めてから判断されたら良いと思います。英語の勉強にもなりますし(字幕もあります)、MITのコース受けてるって行ったらなんか格好いいし!
課金したのに感想のレベルが低すぎてすみません・・・
ちなみに、同じMicroMastersで現在edXで「Fundamentals of Statistics」が開講中です。 こっちはLecture 0からさっぱりわからなくて諦めた・・・。 新しい分野を英語で勉強するのは私には難しすぎました。
GW読書感想文
GW10連休、皆様いかがお過ごしでしょうか?充実したお休みでしたでしょうか? GW期間中もお仕事だった皆様はお疲れ様です。ありがとうございました。
さて、とりあえず創薬レイドバトルがひと段落ついたので関連記事をGithubにまとめなおしました。 レポジトリはこちらです。 ノートブック形式ですが中身はMarkdownというよくわからないファイルですがご笑覧いただければ幸いです。
で、やはりどうにも自分には基本的な知識が足りていない(何がわからないかもわからない状態)ということが分かったので、 とりあえず専門用語を拾うべくいくつか本を読んでみました。 以下、とりとめもない備忘録です。
- 1冊目:コンピュータシステムの理論と実装
- 2冊目: StanとRでベイズ統計モデリング、3冊目: Pythonで体験するベイズ推論
- 4冊目:はじめてのパターン認識
- 5冊目:カーネル多変量解析
- 6冊目:岩波データサイエンス vol.5 スパースモデリングと多変量データ解析
- まとめ
1冊目:コンピュータシステムの理論と実装

コンピュータシステムの理論と実装 ―モダンなコンピュータの作り方
- 作者: Noam Nisan,Shimon Schocken,斎藤康毅
- 出版社/メーカー: オライリージャパン
- 発売日: 2015/03/25
- メディア: 単行本(ソフトカバー)
- この商品を含むブログ (4件) を見る
あれ?そもそもパソコンってどうやって動いているんだろう??みたいなレベルなので、とりあえずコンピューターサイエンスっぽい本を読んでみました。
最も基本的なユニット(電子ゲート)からはじめて、最終的にコンピュータシステムをつくるに至る過程を、一つずつ段階を踏みながら実践していこう!という内容です。 「イントロダクション」に引用されている言葉がこの本のスタンスを最も明確に示していると思うので引用します。
「人に重要な影響を与える唯一の学びは、自己発見・自己本位による学び、つまり、経験によって理解された真実のみである」 by カール・ロジャーズ(心理学者)
実際に作り上げるという経験を通して「コンピュータの動く仕組み」を理解しよう、というのが本書の目的ですが、では具体的にどうすればよいのか? 著者は「抽象と実装」を繰り返すことで可能になると述べています。 複雑なプロジェクトを成し遂げるためには、「抽象化」によりモジュールに分割し、それを組み立て直すというアプローチを取るべきだというのがその主張です。
「どのようにおこなうか(how)」という詳細を無視して、「この要素が何をするのか(what)」だけを考えて抽象化すること。 この段階が重要かつ実例に触れて学ぶべきものなので、「抽象化」は著者が解説・提供し、その実装の経験を読者が積む、という構成になっています。 そのために本書では解くべき課題とともに、導入の容易な実行環境、手順が提供されています。
特に私のような初学者にとって親切だと感じた点は、本書全体としては「ボトムアップでコンピュータをつくりあげる」という構成になっているものの、 最初のイントロダクションで「これから何をおこなおうとしているのか?」トップダウン的にプロジェクト全体の俯瞰を提供してくれている点です。 読み進めていくうちに何度も「あれ?いま何やってんすかね??」と迷子になりかけてましたが、その度にイントロに戻ることで全体の中での位置付けを確認することができました。
と、まるで理解したかのように書いていますが私は1章の論理回路で実装をあきらめました(知的体力が無さすぎる・・・)
とりあえず本書の中の図を切りはりすると以下のような話です。たぶん・・・

NANDからスタートして作って行く話が、パーセプトロンの組み合わせと重み変えていろんな関数表現できるよ、みたいなDeepなお話に繋がっている気がしてちょっと賢くなった気分。(違うか・・・)
2冊目: StanとRでベイズ統計モデリング、3冊目: Pythonで体験するベイズ推論
")
StanとRでベイズ統計モデリング (Wonderful R)
- 作者: 松浦健太郎,石田基広
- 出版社/メーカー: 共立出版
- 発売日: 2016/10/25
- メディア: 単行本
- この商品を含むブログ (10件) を見る

Pythonで体験するベイズ推論 PyMCによるMCMC入門
- 作者: キャメロンデビッドソン=ピロン,玉木徹
- 出版社/メーカー: 森北出版
- 発売日: 2017/04/06
- メディア: 単行本(ソフトカバー)
- この商品を含むブログ (1件) を見る
お正月に「データ解析のための統計モデリング入門」という本をよみました(緑本?)。 統計モデリングについて、簡単なモデルから徐々に複雑なモデルへと、非常にわかりやすく解説されており、各所でオススメされている素敵な本なのですが、 後半、ベイズ統計モデルの話になったところで私は理解があやふやになりました。
- モデルが複雑化し、パラメータが増えるに従ってパラメータの推定が困難になる。(なるほど)
- 多数のパラメータの推定方法としてMCMCサンプリングがある。(・・・ほう)
- MCMCサンプリングで得られる定常分布はパラメータの確率分布(・・・はあ)
- だが頻度主義では「真の値」が1つあるという考えだから分布はあり得ない(・・・それはまた)
- でもベイズ統計モデルは確率分布としてあつかう枠組み(・・・ふーん)
- だから、今考えている統計モデルはベイズ統計モデルだったと見直そう(・・・・・・えっ?)
っていう感じの流れでした。4番目くらいで頭がついていかなくなりました。(3段階が思考の限界なのです。)
しかしベイズという名前はあちこちで見かけるので、やはりお勉強しなければならないのではないか、ということで2冊ほどパラパラめくってみました。
今回読んだ2冊はいずれも実践的な面を重視しており、ベイズモデリングのためのソフトウェアとしてそれぞれ「RとStan」、「PythonとPyMC」が使われています。 コード例が多数記載されていて、特に「グラフの描画」がこまめに挿入されている点が実際の解析過程をなぞっているようで面白いと思いました。
が、しかし、ここまで多数の例を懇切丁寧に紹介されても私の理解力ではしっくりこない。ぼんやりとネットをうろうろしていたところ、以下の解説記事を見つけました。
- 作って遊ぶ機械学習 ベイズ推論による機械学習の基本
下の本の著者の方が書かれているブログのようです。(著書は未読です。すみません。ちらっと見たら数式が多そうで手がでませんでした。)
")
機械学習スタートアップシリーズ ベイズ推論による機械学習入門 (KS情報科学専門書)
- 作者: 須山敦志,杉山将
- 出版社/メーカー: 講談社
- 発売日: 2017/10/21
- メディア: 単行本(ソフトカバー)
- この商品を含むブログ (1件) を見る
上記のブログ記事の「ベイズ学習における基本的な流れ」を読み、やっと書籍で行われていたコードの流れが少しわかるようになりました。引用させていただきます。
- 尤度関数(観測データの表現方法)を定義する
- 事前分布(パラメータの散らばり具合)を設定する。
- ベイズの定理を使って事後分布を更新する。(=学習)
- 事後分布から予測分布を計算し、未知の値の確率分布を求める。(=予測)
こちらを見るまでは、一つ一つのコードを見ているうちに全体として何をやっているか追えなくなる、ということを繰り返していました。*1
流れがわかるようになったところで、改めてベイズ推論についてです。
考え方の導入としては③「Pythonで体験するベイズ推論」の「第1章 ベイズ推論の考え方」が「気持ち」を理解する上でわかりやすかったです。
曰く、ベイズ主義とは「信念」であり、さらに「人によって信念が異なっていることも定義が許している」点が、我々が日常で行なっている推論と類似しており自然な考え方、とのこと。(何を言っているかよくわからないと思うので気になった方は書籍の1章の最初だけでもご覧になっていただければと思います。)
②「StanとRでベイズ統計モデリング」2章のとても簡潔にまとめられた特徴と合わせると以下のような感じです。

もう一つのキーワードMCMCですが、こちらは事後分布を求めるところで出てきます。 複雑なモデルではデータの得られる確率(p(D))を求める計算の難易度が高いため、 事後分布(p(θ|D))に比例する分布 p(D|θ)xp(θ)から乱数サンプルを多数発生させて事後分布の代わりとするそうです。*2
以上のような導入を踏まえて、読者の理解が進むよう、書籍②、③ともに豊富な事例を提供してくださっています。
- 「実際にモデルを作成するにははどのような分布を選択すれば良いのか?」
- 「MCMCの短所として、時間がかかったり収束しないということがあるけど、そんな時どうすればよいのか?」
といった話題に踏み込んで解説してくださっていますので、「頭のいい人はそう言う考え方でアプローチするのか」と非常に興味深かったです。
といってもほとんど理解できていないのでもう一回読んだ方が良さそう・・・。
4冊目:はじめてのパターン認識

- 作者: 平井有三
- 出版社/メーカー: 森北出版
- 発売日: 2012/07/31
- メディア: 単行本(ソフトカバー)
- 購入: 1人 クリック: 7回
- この商品を含むブログ (5件) を見る
「機械学習 教科書」で検索すると必ず出てくる「はじパタ」こと「はじめてのパターン認識」・・・。
重い腰をあげて漸く購入したのですが、私には難しすぎました。本当に皆さんここからはじめているのでしょうか? 分厚すぎず教科書として無駄がない良い本なのだろうなあ、ということは印象ですが、はじめっからハードル高すぎでは・・・???*3
個人的には 第5章 k最近傍法で
- ボロノイ図がでてきた(「CASTpの理論で見たやつだ!」)ところと
- 次元の呪いの説明(「これがサクサクメロンパンの呪いか!」)
がピークでした。
ただ、分からないなりに読み進めていく中で、雰囲気として「統計モデリングのお話と機械学習ではちょっと重点のおいている場所が違う」というのを感じました。
先に読んだベイズ関連の書籍では、背後にあるモデルを意識していることを前提としていました。 一方で「パターン認識」では「今あるデータを分析し、新しいデータの予測を当てること」に重点があり、なんなら 「現実の理由がわからないならそれでもいいんじゃない」くらいの雰囲気を感じました。
これも数式のもつ意味合いを理解できない私の力不足のせいでしょう。いつか理解できる日が来るのでしょうか???
5冊目:カーネル多変量解析
")
カーネル多変量解析―非線形データ解析の新しい展開 (シリーズ確率と情報の科学)
- 作者: 赤穂昭太郎
- 出版社/メーカー: 岩波書店
- 発売日: 2008/11/27
- メディア: 単行本
- 購入: 7人 クリック: 180回
- この商品を含むブログ (32件) を見る
はじパタ「第8章 サポートベクトルマシン」で「カーネルトリック」という言葉を目にし、 「なんか知らんけど響きが格好いい!!」となったので購入しました。(安直)
結論としてこちらの本、門外漢にとってはとても楽しかったのです。なぜかというと全7章のうち、最初の4章が 「カーネル法すごいぞ!」「こんなことできてしまうぞ!」という「ご利益」の説明に重点がおかれており、 証明や理由の込み入った部分については適宜飛ばしつつでも読める、というように書かれているからです。
もちろん残りの後半の章で、前半で触れられなかった数学的な話が展開されています(・・・つまり私は後半を飛ばした)。
第1章ではまず現代の多変量解析においてカーネルがどのような役割を果たすか、その位置づけが語られます。
そもそもカーネル法とは、「カーネル関数の重み付きの和で表したモデルを正則化付きで最適化するデータ解析手法」と言えるそうです。
カーネル法のご利益は以下のような感じです。
- 線形モデルと異なり入力データとして非線形なもの(文字列やグラフ構造でもOK)を取ることができる
- 「カーネル関数の重み付きの和」を扱うので決めるべきパラメータ自体には線形性がある
- サンプル数(入力データ)と同じだけの自由度があるため複雑な関数を表現できる ⇄ 汎化能力が問題となる(正則化で解決を試みる)
- カーネルの計算とその後の処理を分離できるので、計算をモジュール化できる。
次いで第2章では、カーネル法で使われるカーネル関数はどのようなもので、どうやって導入することができるのか、複数の観点から定義が試みられています。
注目の「カーネルトリック」は、「正定値性からの導入」で取り上げられています。
曰く、「正定値性をカーネル関数の定義とすれば」、「内積計算が不要」となり、「計算量をぐっと減らすことができる」そうな・・・。
この一つ前に語られている「特徴抽出からの導入」ではカーネル関数が「特徴ベクトルΦ(x)どうしの内積」として定義されていました。
これに対して「正定値性を満たすことがわかっている関数」を利用すれば、内積計算で導く必要なくカーネル関数を用意できることになるそうです。(・・・???)*4
ここまでの例示は回帰の話題が取り扱われているのですが、第3章、第4章と、より複雑な問題へ話題が移ります。
第3章では固有値問題の例として「カーネル主成分分析」や「カーネル正準相関分析」が、
第4章では凸計画問題の例として「サポートベクトルマシン」や「スパース性」について触れられています。
私はそもそも「カーネル」が接頭語としてついていない「主成分分析」「正準相関分析」もあやふやな段階だったので、「そんなお話もあるのね」程度にしかわかりませんでしたが、だいたい第3章の手順としては、繰り返し
が行われていました。
第4章 SVMの解説はとても面白く、「はじパタ」ではいまいち理解できなかった部分についてもう少し追うことができました。
まず、そもそもの出発点として、クラス分類の識別関数の誤差に対するペナルティの考慮からスタートしています。
問題意識として、
- 損失関数に2乗誤差を用いると外れ値の影響が大きくなる(ロバスト性)
- サンプル数が多すぎる場合計算量が大きくなりすぎる(スパース性)
という点があげられています。これに対して
- 「最小2乗ではない物」を使いたい + 「凸関数」をつかいたい、と言うことで損失関数に「区分線形損失」を導入
- これを解いた解は、サンプルの一部分しか使わないため(スパース)、計算量が節約できる
という解説がなされています。より具体的には、
といった流れです。この中で、
- KKT条件の相補性条件から解のスパース性(サポートベクトルが全体のサンプルに比べてスパースになる)
が導かれるという説明がなされます。書き写しておきながら全然理解していませんが、未来の自分のためのメモです。ご容赦ください。
「それでは、具体的なカーネル関数の中身はどのようなものなのか?」「どう設計すれば良いのか?」という話題は第5章以降に展開されていますが、またしても私の能力では追いきれませんでした。
6冊目:岩波データサイエンス vol.5 スパースモデリングと多変量データ解析

- 作者: 岩波データサイエンス刊行委員会
- 出版社/メーカー: 岩波書店
- 発売日: 2017/02/16
- メディア: 単行本(ソフトカバー)
- この商品を含むブログ (3件) を見る
GW読書感想文、最後ははじめての「岩波データサイエンス」です。 これはもう単純に「スパース性」の話題を知りたかった、というだけです。
結論・・・「えるわん」
なんだかすごくL1でlassoでした。めっちゃ0になるらしいです。
明治大学 データ化学工学研究室 金子先生のページに、 「Lassoの回帰係数がなぜ0になりやすいのか?」について解説が記載されていました。 「係数が0になりやすい」というのを目にし、吝嗇家としては「もったいない・・・」となりましたが、 見方を変えて「特徴選択」という観点からは、「0になることにこそ価値がある」という話になるようです。
その発想はなかった・・・。
フィンガープリントを計算して見た際に、「立っているビットの数が少ない」という印象をうけ、「なんだかスパースを考えないといけないのでは!!!」というのが興味をもったきっかけだったのですが、とりあえずL1正則化項を入れれば解決、ということでしょうか???
まとめ
以上、いつにもましてとりとめもない備忘録でした。 理解できないとはわかっていましたが、ここまでわからないとは・・・。 そもそももっと腰を落ち着けて時間をかけて読むべき本ばかりなのでしょうが、如何せん計画を立てて勉強できない自分の性格をなんとかしないといけなさそうです。
カリキュラムがある大学はいいなあ。
スコア関数を変えてドッキング結果を再解析する話
さて、前回の記事でドッキングによるSBVSもなんとか終わったので 後は化合物IDのリストを提出するだけ・・・、というところですがとても気になる文献を知ってしまいました。
Performance of machine-learning scoring functions in structure-based virtual screening. Wójcikowski, M., Ballester, P.J., and Siedlecki, P., Sci.Rep. 2017(7)46710 pubmed
こちらの文献は前回使用したODDT(Open Drug Dicovery Toolkit)と同一の筆頭著者によるもので、どうやらドッキングのスコアを機械学習を用いて改善したという内容のようです。しかもODDT同様に、このスコア関数(RF-ScoreVS)を利用できるとのこと!!
前回の記事で参照した「タンパク質計算科学 基礎と創薬への応用」*1 において、ドッキングのスコア関数は依然として精度に課題がある(実験値との乖離がある)といった趣旨の記述がありました。AutoDock VinaはAutoDockよりも精度が改善している、とはいえ10年ほど前のソフトウェアです。より改善したものがあるのであれば使ってみたい!
(・・・というのは建前でMachine Learningって言って格好つけたかった)
1. RF-Score-VS (Sci.Rep. 2017(7)46710)文献紹介
まずは文献を読んでみようと思います。Scientific ReportsはOpen Accessなのが良いですね。(CC BY 4.0)
すでに記憶があやふやなのでスコア関数について少し復習します。
1-1. スコア関数の種類
スコア関数にはどのようなものがあるか?
先の記事で参照した日本化学会情報化学部会誌の文献( タンパク質-リガンドドッキングの現状と課題*2 によると、大きく3つ
- ①力場に基づいたスコア関数(force-field-based scoring function)
- ②経験的なスコア関数 (empirical scoring function)
- ③ 統計に基づいたスコア関数(knowledge-based scoring function)
があり、さらに近年の動きとして、
- ④複数のスコア関数を利用するコンセンサススコア(concensus scoring)
- ⑤機械学習に基づいたスコア関数(machine-learning based scoring function)
が開発されている、とのことでした。
「④ コンセンサススコア」について「タンパク質計算科学」の説明を参照します。
「コンセンサス(consensus、合意)」をとる目的は、単一の手法の結果のみに頼らず、複数のスクリーニング結果を統合することでヒット率の向上、当たり外れの低減を目指すことです。結果を統合する方法として、各化合物に対して複数の手法でスコアを得たのち
- 積集合: 複数の手法で共通に選ばれた化合物をヒットとする
- 和集合: 複数の手法で少なくとも一つに選ばれた化合物をヒットとする
- rank by scoring: 各手法のスコアを「0~1」の範囲にスケールした上で和をとり、新しいスコアとする
- rank by rank: 各手法における化合物の順位(ランク)の平均によってランキングする
といった手法が例に挙げられていました。
では、「⑤ 機械学習に基づいたスコア関数」はどのようなものか? 論文の中身に進みます。
1-2. 既存のスコア関数の問題点
関数の構造上の問題
そもそもなぜ機械学習の導入にいたったか? 論文中では従来から用いられているスコア関数を"classical scoring functions (SFs)" (おそらく「①力場」や「②経験的」、「③統計」によるスコア関数のこと?)としており、結合親和性の予測精度の向上に限界が見られていることの理由として、以下の問題点を指摘しています。
- コンフォメーションのエントロピーをうまく説明しない
- 溶媒和によるエネルギーの寄与をうまく説明しない
また、従来のスコア関数は線形回帰モデル(linear regression model)を下敷きにしているため、
- 大量のタンパク質構造や結合に関する情報を吸収してモデルに取り込むことに限界がある(universal SFの限界)
- 標的のタンパク質(protein family)に焦点を合わせてスコア関数のパラメーターをチューニングしたくても、回帰モデルが変更できる形では提供されていない
といった問題を指摘しています。・・・難しい。
よくわからないので別の文献のFigureを引用します。(WIREs Comput. Mol. Sci.2015(5)405)*3

Fig 中に示されているようにClassocal SFでは線型結合の式ですが、Machine-learnig SFでは関数となっています。そもそもモデル化の構造が異なっているということで、異なるパフォーマンスが期待できるよね!ということのようです。
モデル作成上の問題
さらに、classical SFのモデル作成時の問題点として、トレーニングセットとテストセットに重複(overlap)があるということが挙げられています。機械学習であればcross validationにより、より厳密なバリデーションの取り扱いが可能になります。
また、classical SFは結晶構造の情報を元に作成されていますが、これは結合するというプラスのベクトルの事例のみの情報を用いて構築していることになります。一方でスコア関数を適用するVirtual Screeningの目的は、スクリーニング結果として得られた「実際に結合する正例(プラスのベクトル/ +ve)」と「結合しない負例(マイナスのベクトル/ -ve)」が入り混じった状態である一連の結合ポーズの中から、正しいtrue binderを見つけ出すということです。つまり、Virtual Screeningでは負例(inactiveのリガンド / -ve)の比率の方が高いのに、その情報が関数作成段階に考慮されていないと指摘されていました。
(なるほど確かに言われてみればそう・・・なのでしょうか?勉強不足のため十分に理解できていません。)
ということで、機械学習を導入することで、これらの問題点を解決して行きましょう!
1-3. 著者らによる機械学習スコア関数(RF-Score シリーズ)
文献中では機械学習スコア関数の例としてRF-Score、NNScore、SFCscoreといった名前が挙げられています。このうち、RF-Scoreは今回の文献の著者の一人、De. Pedro Ballester(CRCM)が筆頭著者として2010年に報告しているものです*4。RFはRandom Forestの略のようですが、こちらをversion 1 (RF-Score v1)として、RF-Score v2*5が報告されています。特にRF-Score v3の文献はタイトルが「Improving AutoDock Vina Using Random Forest: The Growing Accuracy of Binding Affinity Prediction by the Effective Exploitation of Larger Data Sets.」、とAutoDock Vinaを使った以上とても気になる文献です(アクセスできなかった・・・)。
これらのスコア関数の提案に対する議論、「overfittingしているのでは?」「新しいターゲットにも適用できるの?(applicablity)」に対して、非常に大きなデータセットを使って学習を実施し、virtual screeningのパフォーマンスが上げられることを示した、というのが2017年のSci. Rep.の概要となります。
1-4. RF-Scoreの仕組み
では、ベースとなったRF-Scoreはどのようなものなのか?ちょうど良いスライド資料があったので参照していきたいと思います*6。(元文献を読む能力がなくてすみません)
RF-Scoreではランダムフォレストを使ってスコア関数をモデル化していますが、元々の発想としては"classical SF"のうちの「3. 統計に基づいたスコア関数(knowledge-based scoring function)」を改善する、ということから始まっているようです。
統計的スコア関数の流れは以下のようになっています。
- ①多数の結晶構造(PDBのタンパク質-リガンド複合体構造(P-L complex))を比較
- ②原子(あるいは官能基/フラグメント)にタイプを割り当てる
- ③中心の原子と周囲の原子のtype-type間の距離のヒストグラムを作成する(存在確率の空間分布)
- ④存在確率(P)を結合自由エネルギー(ΔF)に変換する(ΔF = -kBT lnP)
- ⑤全てのP-Lアトムペアのエネルギーを合計して全体のエネルギーとする。
上記の「④ヒストグラムのエネルギー関数への変換」では"reverse Boltzmann"の手法が使われています。ここで、「温度Tの平衡状態においてタンパク質とリガンドの各原子は独立な粒子である」という仮定が入りますが、これは仮定としては粗すぎる、というのが問題となるようです。
理由として
- 分子中の原子と原子は結合している(Molecular connectivity)から、原子と原子の距離は独立とはいえない
- 排除体積の影響(Excluded volume)
- 平衡状態の仮定に物理的根拠がない
- 温度(T)による構造変化は小さく、ボルツマン分布により暗に考慮されているほどではない
といったことが挙げられています。 以上をふまえて、著者らは「ヒストグラム(距離の分布 / distance distribution)と結合自由エネルギーとを関係付ける」ために、Boltzmannの公式のような公式を仮定するのではなく、「構造情報と結合親和性の関係性を機械学習を使って学習すれば良い」と考えたのがRF-Scoreの出発点となっているそうです。
上記の考えに基づき、以下の操作を行って作成したモデルがRF-Score (v1) となります。
- ①PDBの構造情報(PDBbind Benchmark)と質の高い結合親和性データ(Kd, Ki)
- ②タンパク質とリガンドの、閾値以下(dcutoff)の距離にある原子をみつける
- ③アトムタイプペアをフィーチャーとして出現率を求める
- ④各タンパク質-リガンド複合体をフィーチャーのベクトルとして表す
- ⑤ベクトルを入力(説明変数)、結合親和性を出力(目的変数)としてランダムフォレストによる機械学習を実施
加えてさらに、スコア関数の精度向上にむけて公共データと様々な製薬企業の企業内データを用いてスコア関数の精度向上を目指したコンソーシアム(Scoring Function Consrtium)のデータに基づきフィーチャーの選択を改良した(RF-Score v2 / Data-driven feature selection)や、AutoDock Vinaのフィーチャーを取り入れる(RF-Score v3)、といった改良を加えてモデルの精度を高めてきた、というのが流れとなります。
1-5. RF-Score-VSでの取り組み
先に述べたようにRF-Score-VSでは、学習のデータセットに工夫を加えることでVirtual Screeningにおけるスコアの改善が目指されています。
データセットの工夫
まず、データセットとしてDUD-E(A Database of Useful Decoys: Enganced)データセットが使われています。DUD-E*7はスクリーニングソフトの性能テスト(ベンチマーク)として利用されているDUDの改良版であり、DUDは市販化合物の3次元データベースであるZINCのデータを既知の標的タンパク質ごとに、その既知の活性化合物(active)と、類似した化合物(inacitive)との組み合わせとして整理したデータセットであるとのことです*8。
先述のように、RF-Scoreの学習セットとしてはPDBの情報が用いられていました。これに対してRF-Score-VSの利用している学習セットには
- より大きなデータセット
- 結晶構造ではなく、active リガンド(+ve)とinactive リガンド(-ve)のドッキングポーズ
を利用するという違いがあります。結晶構造はactiveリガンドのポーズしか含まないので、こちらを学習セットとして用いるよりも、activeとinactive両方の結合ポーズを学習データとして用いる方がVirtual Screeningに用いるスコア関数としてはより実際の使用状況に類似しているだろう、というのが背景にあるようです。
学習データ分割方法の工夫
また、興味深いことに学習データセットのcross-validationを行うにあたって、
- ①標的タンパク質ごと(per-terget)
- ②訓練データ、テストデータともに全ての種類のタンパクを含む(horizontal split)
- ③訓練データとテストデータで、標的タンパク質が重複しないようにした上で分割する(vertival split)
という3種類の分割方法が用いられています。
論文中の図を引用します。

②horizontal splitは「すでに活性化合物(active ligand)が知られている標的についてvirtual screeningを行う」という状況を、③vertical splitは「既知の活性化合物がない状態でvirutal screeningを行う」という状況を模倣する、というのを目的としているそうです。
Virtual Screeningが行われる状況の場合分けを想定して、訓練データとテストデータの分割方法を分けるというのは、なるほど〜という感じでとても面白いと思いました(・・・よく使われる方法なのでしょうか?)。
1-6. RF-Score-VSの性能
評価指標
評価指標(metrics)としてはEnrichment factor(EF)というものが用いられています。
$$ EF_{x\%} = \frac{\frac{ActiveMolecules(x\%)}{Molecules(x\%)}}{\frac{ActiveMolecules(100\%)}{Molecules(100\%)}} $$
ランク付けした化合物の上位x%を取り出して、その中に本当の活性化合物が含まれている割合を求めます。この割合と、全化合物(x = 100)中に含まれる活性化合物の割合の比をとったものがEFとなります。Virtual Screeningの性能指標としてよく用いられるものには、EF以外にROC(Receiver Operating Characteristc)曲線のAUC(Area Under the Curve)があるそうです。
EFを用いた理由として、Virtual Screeningを行って選んだ化合物を実際にアッセイするとしたら、上位数パーセントを選んで行うことになる。その数パーセントに"当たり"が含まれていることが大事なので、その状況をより反映しているEFの方が適している、といった説明がなされていました。
確かに創薬レイドバトルも「スコアでランキングをつけて上位10個をアッセイ候補として提出」ということになっていました。この論文とても勉強になる。
性能
ではAutoDock Vinaをドッキングに用い、得られた結合ポーズを各種スコア関数でスコアリングして比較した結果を引用します。横軸が Enrichment Factor 1% (EF1%)で、縦軸にスコア関数が並んでいます。

分割方法として② horizontal splitを用いた、RF-Score-VSに置いて非常に良い結果が得られており、従来のclassical SFと比較して高いEF1%となっています。① per-targetは平均値としては、性能が向上しているものの、標的によって性能のばらつきが大きいという結果になっています。また、③ vertical splitは ② horizontalよりも性能は落ちていますが、classical SFよりも性能が向上していると記載されていました。
AutoDock Vinaのスコア関数を改善したという「RF-Score v3」が上記の結果では「AutoDock Vina Score」と比べて良くなっているように見えないのが気になる点ですが、評価対象のデータセットの質が変わった(結晶構造 → ドッキングポーズ)ことが理由なのかもしれません。
他のドッキングソフト(Docking engine)を使った比較結果、DUD-Eとは異なるデータセット DEKOIS 2.0 に対して適用した際の結果についても記載されていましたので、ご興味のある方は論文を参照していただければと思います。
要するに、とにかく良い結果が出るっぽい!!! 使いましょう!
2. RF-Score-VS を使ってみる
2-1. ドッキングスコアを再計算
とても有り難いことにRF-Score-VSに関して、データセット( http://github.com/oddt/rfscorevs )、学習済みのモデル (http://github.com/oddt/rfscorevs_binary )ともにGitHubで公開されています。
各OS向けの学習済みのbinaryが用意されているので、ready-to-use RF-Score-VSをローカルに落として来ました。
こちらのbinaryにはRD-SCORE-VS v2 が入っている、とのことです。
使い方は簡単で、ターミナルでダウンロードして来たフォルダ(rd-score-vs_v1)に移動します。
以下でヘルプを参照できます。
$ ./rf-score-vs --help
ドッキングに使ったタンパク質のpdbファイル(今回は5nix_chainAB.pdbという名前)と、AutoDock Vinaのドッキングで得たリガンド構造を含むsdf(ODDT_Dock_All_Result_mod.sdfという名前)を同じフォルダに入れてコマンドを実行します。
./rf-score-vs_v1 --receptor 5nix_chainAB.pdb ODDT_Dock_All_Result_mod.sdf -O ODDT_All_ligands_rescored.sdf
引数 --receptor に続いてpdb、sdfを入力し、 -O で指定したファイル名で、再計算されたスコア(フィールド名: RFScoreVS_v2)が書き込まれたsdfが出力されました。
今回は全てのドッキングポーズ(50化合物 ポーズ合計449個)についてスコアを再計算しています。では結果を確認していきましょう。
2-2. スコアを確認
RDKitでSDFを読み込んでおきます。
from rdkit import Chem from rdkit.Chem import AllChem, PandasTools import pandas as pd #PandasToolsで読み込み、スコアのTypeをfloatに変換する Rescored_df = PandasTools.LoadSDF('./ODDT_All_ligands_rescored.sdf') Rescored_df['RFScoreVS_v2'] = Rescored_df['RFScoreVS_v2'].astype(float) Rescored_df['vina_affinity'] = Rescored_df['vina_affinity'].astype(float)

AutoDock Vinaのスコアは結合自由エネルギ-の予測値(kcal/mol)で与えられるのに対し、RF-Score-vsのスコア再計算結果は pKd/i の単位で与えられます。
学習の段階でDUD-Eの化合物の活性を pKd/i = 6 よりも弱い(小さい)場合にinactiveとしています。また、トレーニングセットのDecoy(構造類似の不活性化合物?)には一律に pKd/i = 5.95 を設定しています。従って予測値が6よりも大きければ活性あり、となりそうです。
スコアの分布をみてみましょう。ヒストグラムを描きます。
import matplotlib.pyplot as plt %matplotlib inline plt.hist(Rescored_df['RFScoreVS_v2'], bins=20, range=(5.8, 6.8))

要約統計量を確認します。
Rescored_df['RFScoreVS_v2'].describe() """ count 449.000000 mean 6.005943 std 0.075275 min 5.950000 25% 5.960443 50% 5.977587 75% 6.023838 max 6.438919 Name: RFScoreVS_v2, dtype: float64 """
最小値は 5.95で、inactiveのスコア設定にあった値が出ています。第1四分位数 5.96、第3四分位数 6.02と、3/4近くの結合ポーズが pKd/i=6以下となっています。活性値の判断基準(6以上)からすると、活性なしと判別されたポーズが 3/4 近いということになりそうです。(・・・この解釈で良いのだろうか??)
AutoDock Vinaのスコアと一緒にプロットしてみます。
x = Rescored_df['RFScoreVS_v2'] y = Rescored_df['vina_affinity'] plt.scatter(x,y) plt.title("RF-Score-VS vs AutoDock Vina") plt.xlabel("RF-Score-VS") plt.ylabel("AutoDock Vina") plt.grid(True)

AutoDock Vinaのスコアは小さいほど良く、RF-Score-VSは大きいほど良い結果、となりますので左上から右下に向かう対角線方向に動いて入れば、予測結果に相関があることになりそうです。
図を見るとRF-Score-VS (x = 5.5, inactive)の線上に様々なy値の点が分布ししており、今回の再スコアリングではかなり異なるスコアリング結果が得られていそうです。特に興味深い結果なのが、y軸で最小値に近い点(前回記事でスコアが良いとして選択していた点)が、再スコアではかなりスコアが悪いという結果になることです。こんなに違う結果になるとは。。。
2-3. 化合物ごとのベストポーズを取得
上のプロットは全499個の結合ポーズ全てなので、ここから各化合物ごとに最も良いスコアとなっているポーズのみを取り出したいと思います。元々の化合物のIDを「original_id」というフィールドに格納しているので、これを使ってgroupbyすれば良さそうです。
# 各化合物ごとのポーズ数を確認してみる Rescored_df.groupby('original_id').size() """ original_id PB27242793 9 PB300184784 9 PB57131569 9 PB90021090 9 ・・・以下略 """
len(Rescored_df.groupby('original_id').size()) #50
original_idでグループ化したものから、RFScoreVS_v2の値が最大となっているものを取り出して新しいデータフレームとします。*9
id_group = Rescored_df.groupby('original_id') best_scores = Rescored_df.loc[id_group['RFScoreVS_v2'].idxmax(), :] # 化合物数を行数で確認 best_scores.shape # (50, 18)
再度、二つのスコアでプロットしてみます。(コード省略)

だいぶんスッキリした見た目になりました。要約統計量を確認します。
best_scores['RFScoreVS_v2'].describe() """ count 50.000000 mean 6.079934 std 0.111637 min 5.967962 25% 6.004503 50% 6.054512 75% 6.096116 max 6.438919 Name: RFScoreVS_v2, dtype: float64 """
今回は第1四分位数で6.00、第2四分位数で6.05と、各化合物のベストスコアを抜き出したことで安心できる分布になりました。これなら希望が持てるかもしれません・・・
プロットに合わせて構造を確認してみたいと思います。@iwatobipen先生の記事Plot Chemical space with d3js based libraryを参考にさせていただき、d3jsを使います。
import mpld3 from mpld3 import plugins mpld3.enable_notebook() #描画を作成するためのモジュール from rdkit.Chem.Draw import rdMolDraw2D from IPython.display import SVG #svgを格納するための空のリスト svgs_list = [] for i in range(len(best_scores)): view = rdMolDraw2D.MolDraw2DSVG(300,300) ROMol_col = best_scores.columns.get_loc('ROMol') mol = best_scores.iloc[i, ROMol_col] prepro_mol = rdMolDraw2D.PrepareMolForDrawing(mol) view.DrawMolecule(prepro_mol) view.FinishDrawing() svg = view.GetDrawingText() svgs_list.append(svg.replace('svg:,','')) x = best_scores['RFScoreVS_v2'] y = best_scores['vina_affinity'] fig, ax =plt.subplots() ax.set_xlabel("RF-Score-VS") ax.set_ylabel("AutoDock Vina") points = ax.scatter(x, y) tooltip = plugins.PointHTMLTooltip(points, svgs_list) plugins.connect(fig, tooltip)

gifではわかりにくいですが、スポットにカーソルを合わせると構造式が表示されます。
2-4. RF-Score VSにおけるスコアTop 10の取得
Top 10を取り出してSDFとして書き出しておきます。
#スコアでソートし、上から10個を取り出す best_scores_10 = best_scores.sort_values('RFScoreVS_v2', ascending=False).iloc[:10] PandasTools.WriteSDF(best_scores_10, 'RF-ScoreVS_Top10.sdf', properties=best_scores.columns)
描画します。前回と同じなのでコードは省略しますが、レジェンドのRFがRF-Score VSのスコアを、ADがAutoDock Vinaのスコアを表します。

AutoDock Vinaのスコアで選択したTop 10の結果と並べてみます。

両スコア関数で選択される化合物の違いを見てみてると、ぼんやりとですがRF-Score-VSではリガンドの中心付近にヘテロ芳香族環が位置していたり、AutoDock Vinaによる選択と比較して、分子全体が丸まっているように見えるものなどもあります。
3. アッセイ候補提出ファイルの作成
新しいスコア関数 RD-Score VSによる再スコアリングと結果の取り出しが完了しました。AutoDock Vinaのスコアリングとそれぞれ違いがあって面白いので、各々Top 5を取り出して合わせて10化合物とし、アッセイ候補としたいと思います。(ダイバーシティー!)
提出フォーマットはタブ区切り、ということなのでcsvモジュールを使ってファイルを作ります。まずはリストを作成。
top_10_list = [] # RFScoreVSを基準に5個取り出し RF_sup = Chem.SDMolSupplier('./RF-ScoreVS_Top10.sdf') for i in range(5): temp_list =[] mol = RF_sup[i] rank_num = i + 1 compound_id = mol.GetProp('original_id') score = mol.GetProp('RFScoreVS_v2') temp_list = [rank_num, compound_id,score] top_10_list.append(temp_list) # AutoDock Vinaを基準に5個取り出し ADVina_sup = Chem.SDMolSupplier('./SBVS_Top10.sdf') for i in range(5): temp_list =[] mol = ADVina_sup[i] rank_num = i + 6 compound_id = mol.GetProp('original_id') score = mol.GetProp('vina_affinity') temp_list = [rank_num, compound_id,score] top_10_list.append(temp_list)
10化合物についてIDとスコアの組み合わせのリストのリストが作成できたので、タブ区切りのファイルを書き出します。
import csv with open('./top10_magattaca.txt', 'w') as f: writer = csv.writer(f, delimiter='\t') writer.writerows(top_10_list)
念のため読み込んで確認
with open('./top10_magattaca.txt') as f: print(f.read()) """ 1 Z393761442 6.438919 2 Z243276918 6.404471 3 Z200290200 6.386205 4 Z368242106 6.385228 5 Z278994950 6.207442 6 Z237879862 -10.9 7 Z19455181 -10.9 8 Z872606870 -10.8 9 Z2788907736 -10.4 10 PB90021090 -10.4 """
無事目的のファイルが作成できていそうです。果たしてこの中に活性のあるものはあるでしょうか???
同様に、500化合物のリストも同じファイル形式としておきます。スコアとしてはLBVSを行なったときの類似性評価のTanimoto係数によるスコアを記載しておきます。 (色々こねくり回して494個と500個に少し足りませんがご容赦いただけると信じて)
Top500_sup = Chem.SDMolSupplier('./SCR_compounds_SimScore.sdf') top_500_list = [] rank_num = 0 for mol in Top500_sup: rank_num +=1 compound_id = mol.GetProp('original_id') score = mol.GetProp('Similarity_Score') temp_list = [rank_num, compound_id,score] top_500_list.append(temp_list)
完了! これを提出フォームに登録すればOK!
4. まとめ
今回はODDTに関して調べているうちに行き当たった論文、Sci.Rep. 2017(7)46710 に魅かれて機械学習を用いたドッキングスコア関数について調べてみました。実際に適用して見るとAutoDock Vinaによるスコアリングと異なる傾向が見え、とても面白い結果だと感じました。文献中ではまだまだ修正・改善が必要だ、と書かれていましたが、既存のスコア関数のモデルの問題点など、非常に学ぶことが多い論文だと思いました。
前回の記事でドッキングを行なった際には、とりあえず動いてデータが出れば良し!ということで進めていきましたが、AutoDock Vinaが少し古いソフトウェアなのかな?これだけで良いのかな?という懸念がありました。ドッキングそのものは時間がかかるので、何度も条件を変えて試すのは大変ですが、結合ポーズが出ていればスコア関数を修正して評価精度をあげることができる、というのを学べて良かったです。(新しいっぽいことをやって格好つけたかった)
とりあえずデフォルト+α のことが少しできたので満足!
5. 総括
提出も完了したので、最後にこれまで行なってきた処理手順をまとめます。
- 前処理(脱塩、構造標準化)
- 指標による絞り込み(主にRule of 5の適用)
- 部分構造による絞り込み(オルト置換ビフェニル構造)
- ファーマコフォアスクリーニング(ファーマコフォアを変えて2回実施) ➡︎ 化合物 Top 500 (厳密には494個)
- LBVS(既知活性化合物に対するフィンガープリントベースの類似性評価による順位づけ) ➡︎ 化合物 Top 50を次工程へ
- SBVS (AutoDock Vina + RF-Score VS) ➡︎ 化合物 Top 10をアッセイ希望に選択
以上の6ステップです。書いてしまうとこれだけ、という感じもありますが、各工程でびっくりするくらいに行き詰まったので予想以上に時間がかかってしまいました。(当初、3月上旬提出だったのに、ずるずる遅れてしまいました。遅れている上に余計な寄り道ばっかりしてしまいましたし・・・。締め切りを4月中に延ばしてくださった創薬ちゃんに感謝です。)
展望も戦略もないままはじめて右往左往していましたが、行き詰まるたびにTwitterで解決方法や、方針、文献諸々を一からご教示いただき、なんともありがたい気持ちでいっぱいです。この場を借りて皆様にお礼申し上げます。今後ともよろしくお願い申し上げます。(図々しくてすみません)
あとは、選んだ化合物の中に活性を持つものがあることを祈って!果報は寝て待ちましょう。
今回の記事も間違いがたくさんあると思います。ご指摘いただければ幸いです。
*1:タンパク質計算科学 基礎と創薬への応用 神谷成敏・肥後順一・福西快文・中村春樹 著 共立出版 2009年初版1刷
*2:Uehara, S., Tanaka, S., CICSJ Bulletin 2016(34)10
*3:Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening Ain, Q.U., Aleksandrova, A., Roessler, F.D., and Ballester P.J. WIREs Comput. Mol. Sci.2015(5)405
*4:A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Ballester, P.J. and Mitchell., J.B.O., Bioinformatics. 2010(26)1169
*5:Does a more precise chemical description of protein-ligand complexes lead to more accurate prediction of binding affinity? Ballester, P.J., Schreyer, A., Blundell. T.L., J. Chem. Inf. Model. 2014(54)944)、RF-Score v3 ((Li, H., Leung, KS., Wong, M.H., Ballester P.J., Mol. Inform. 2015(34)115
*6:ML to improve scoring functions binding and virtual screening
RF-Score: a Machine Learning Scoring Function for Protein-Ligand Binding Affinities
*7:Mysinger,M.M.,Carchia,M.,Irwin,J.J.,Shoichet,B.K.,J.Med.Chem.2012(55)6582)
*8:「タンパク質計算科学」の記述参考