OpenMMをステップバイステップで 〜 Part 1:GUIでタンパク質の前処理 〜
MD計算について調べてみるとオープンソースツールがたくさんあることがわかってきました。ここではGUIが用意されていて、初学者に優しそうなOpenMMでちょっとずつ遊んでみたいと思います。
この記事では第一歩、PDBから取得したタンパク質構造をMD計算にあわせて前処理する方法を試してみます。具体的にはOpenMM SetupでGUIによる処理をおこないます。次回の記事で、同じ処理をPython経由で行う方法をPDBFixerでためします。*1

1. この記事の目標
最後の最後にやりたいことはTeachOpenCADDのT019 · Molecular dynamics simulationとT020 · Analyzing molecular dynamics simulations で行われているようなタンパク質-リガンド複合体のMDシミュレーションと結果の解析を理解することです。
TeachOpenCADDはとても分かりやすい教材ですが、私のようなレベルの低い初学者にはそれでも少し要素が多すぎてついていけないです。もっともっと初歩の初歩から・・・
ということで、まずはMD計算に合わせたタンパク質構造の前処理を試します。TeachOpenCADDと同じ構造(PDB id: 3POZ)を題材にしますが、ここではタンパク質のみでリガンドは考慮しません。
この記事では、まずOpenMMに付随するGUIツールのOpenMM Setupで前処理の流れを確認します。
2. 準備
この記事で使うツールはOpennMMとOpenMM Setupです。すべてcondaがあれば簡単にインストールできます。
conda install -c conda-forge openmm conda install -c conda-forge openmm-setup
計算対象の構造は「タンパク質EGFRのキナーゼドメインとリガンドTAK-285の複合体(PDB ID: 3POZ)」です。

出典となっている論文はこちら
Structural Analysis of the Mechanism of Inhibition and Allosteric Activation of the Kinase Domain of HER2 Protein. Aertgeerts, K., Skene, R., Yano, J., et. al., (2011) J Biol Chem 286: 18756-18765 DOI: 10.1074/jbc.M110.206193
Open Access, CC BY 4.0
3. どうして前処理が必要なの?
そもそもどうしてPDBの構造をそのままシミュレーションにかけるのは要注意なのでしょうか?PDBFixerのマニュアルのイントロダクションが分かりやすいです。

このような問題に対してPDBFixerでは以下のような処理が可能になっています。

これからOpenMM SetupをつかったPDBの修正方法をみていきますが、GitHub : openmm-setupをご覧いただけばわかる通り、裏側ではPDBFixerが走っています。
4. OpenMM Setup で前処理しよう
それでは早速、OpenMM Setupを使った前処理をためしてみましょう!
処理の全体の流れは以下です。合わせて処理の前後の構造を描画しています(PyMol)。
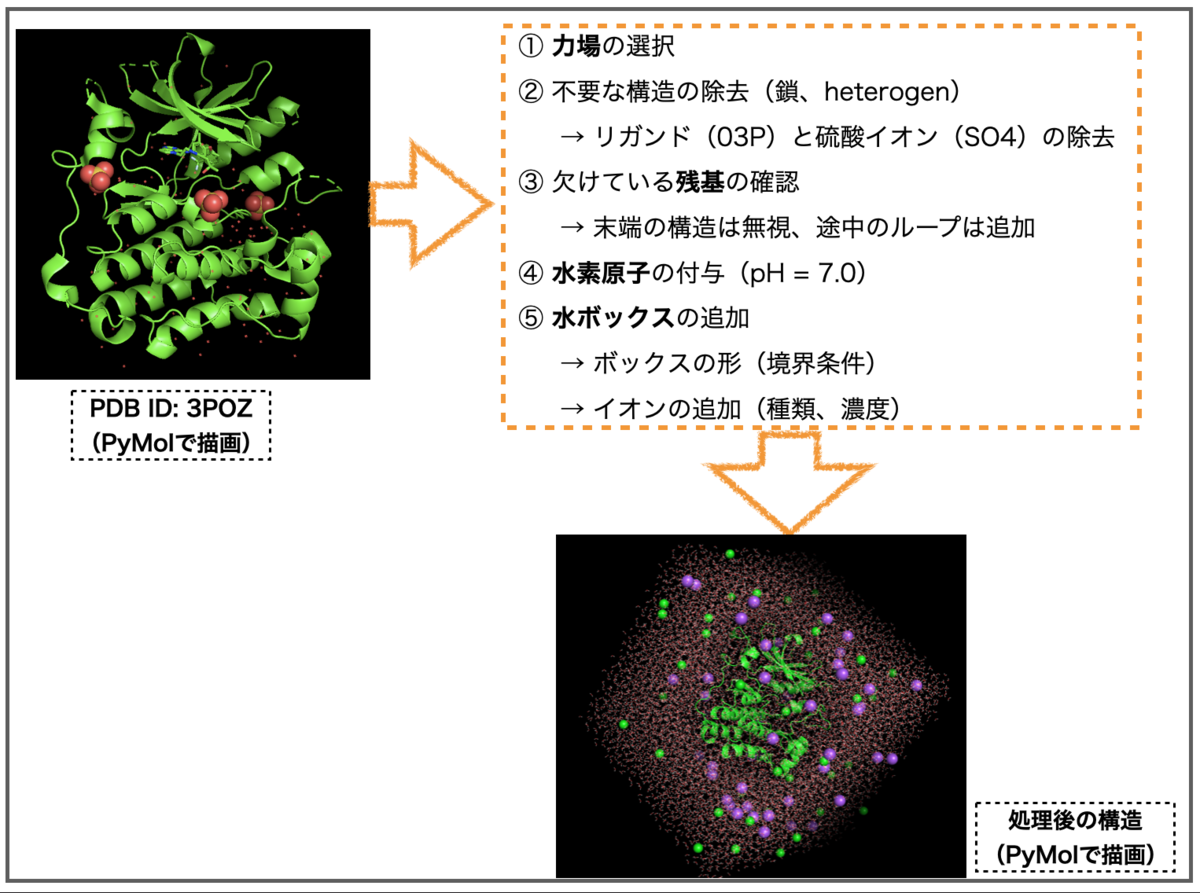
4-1. アプリの起動とファイルの読み込み
OpenMM Setupアプリをcondaでインストールした場合は、仮想環境をアクティベートして以下のコマンドでブラウザが立ち上がります。*3
openmm-setup

PDBファイルを扱うのでそのままcontinueします。
次の画面では① PDBファイルの読み込みと、② 力場の選択、③ 水モデルの選択を行います。Browseをクリックして入力ファイル(Input File)を選択できます。

力場(Force Field)と水モデル(Water Model)はプルダウンから選べます。選択肢を上図右側に貼りました。
ここではTeachOpenCADDに合わせてAMBER14とTIP3P-FBを選択しています。違いはよくわかりません。。。
上図下部では「入力ファイルに修正が必要か否か?」を聞かれます。Yesを選んで修正を加えていきましょう!
4-2. 不要な構造の除去
ついでPDBファイルの構造からシミュレーションの対象にしたい部分を取り出します。複数の鎖(chain)が含まれる場合やリガンド・塩などが入っている場合があるので、不要な構造を除去します。
以下のような画面で設定します。右側の3Dモデルはインタラクティブ表示で、ぐりぐり回転したりズームしたりできます。便利!
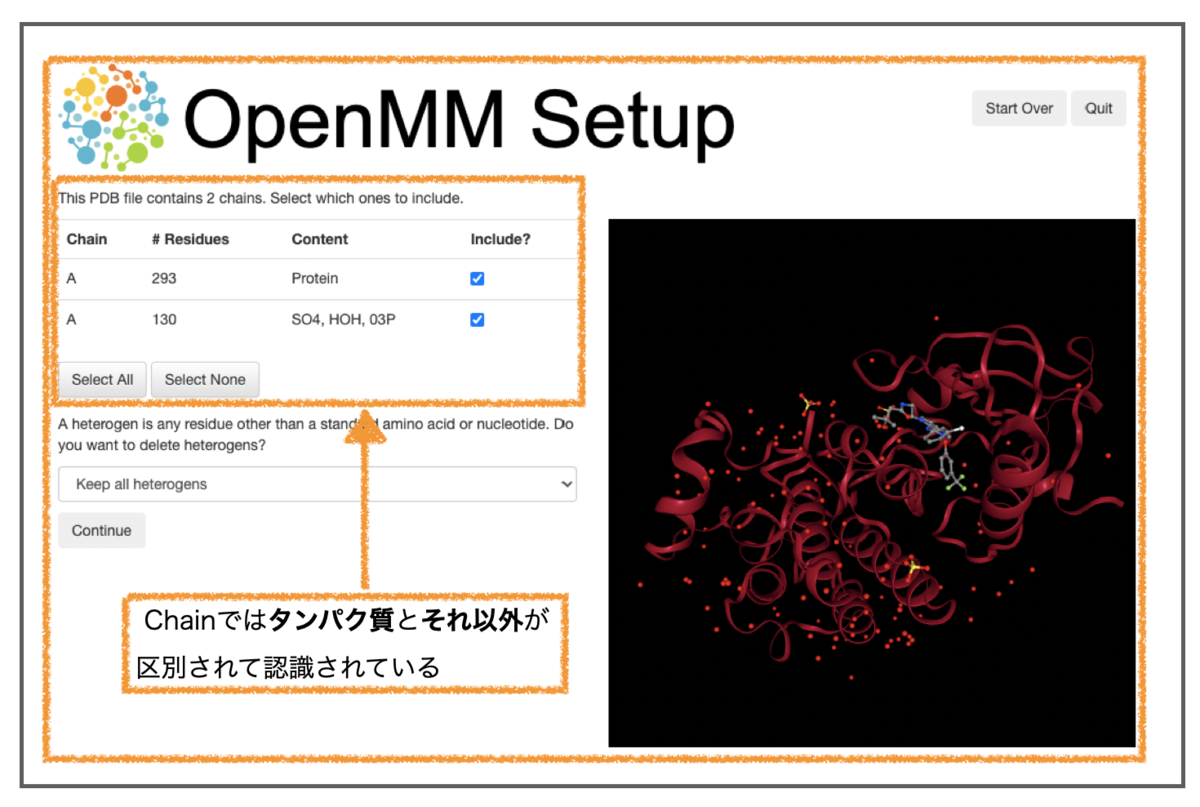
左側上部では鎖の選択ができます。Contentの部分を見るとProteinとそれ以外(SO4、HOH、03P)が区別されているのがわかります。テーブル右端のincludeのチェックの有無で構造を含めるかどうか選べます。
今回のシミュレーションでは硫酸イオン(SO4)とリガンド(03P)を除いた、タンパク質と水(HOH)を対象とします。上部テーブルではProtein以外の分子を水を含めてまとめてあつかっているため、チェックを外すとHOHも消えてしまいます。
そこで上部はこのままにして、下部のHeterogenの設定箇所を使います。

上図のようにDelete heterogens except waterを選択します。これで水を残したまま、それ以外のイオン、リガンドを削除できます。設定の変更は右側3Dビューにも反映され、リガンドなどが消えていることがわかります。
確認できたらContinue!!
4-3. 欠けている残基の確認と追加
続いてタンパク質構造で欠けている残基(missing residue)の確認を行います。以下のように残基の位置と種類を確認することができます。

残基を追加したい時は右端のAdd?にチェックを入れます。今回は両末端は加えず、途中の欠けている残基は追加することにします。(鎖が途中で切れたままだと変な動きになりそうなので・・・)
UniProt KB:P00533(EGFR_HUMAN)等と比較すればわかりますが、「PDB ID:3POZ」のカバーする残基番号は「696 - 1022」です。なので上図でチェックを入れていない「696 to 700」「1018 to 1022」がN末端とC末端に相当します。
参考までに欠けている残基の箇所を確認してみましょう。RSCB PDBサイトの3D Protein Feature Viewが非常にわかりやすいです。

上図左側の「UNMODELED」とがシークエンスの中でモデル化されていない部分です。missing residueはここを見れば良さそうです。
右側の3Dビューでピンク色にハイライトされたシート構造はUNMODELEDな領域に挟まれた2次構造(secondary structure)です。このシート構造につながる箇所が点線で表示されています(水色の矢印)。残基が欠けている部分は点線で仮につながれているようですね。
3D構造とシークエンス表示を対応させてみた感じループやタンパク質の両末端といった箇所が欠けているように思われます。フレキシブルで電子密度がうまく観察できなかったのでしょうか?
なお、上の「3D Protein Feature View」は以下のようにたどると見られます。

4-4. 重原子の欠けた残基の確認
「欠けている残基」につづいて、見えている残基の中で「一部の重原子が欠けているもの」の確認画面に遷移します。
以下の通り、1017番目のロイシン(Leu)残基でOXT原子が欠けています。

OXTはC末端の酸素原子(の負電荷を帯びた状態)を表す標準的な名前だそうです。一つ前のステップで確認した通り、この構造では1018番目以降の残基が欠けています。(本来の)C末端の構造が見えていないため、1017番がPDBファイル末端構造になっているようです。
この画面は「欠けている原子を足すよ!」っていうだけなので、このままContinueして良さそうです。
4-5. 水素原子の付与
ついで、水素原子を付与します。どのようなpH条件の設定の下で水素原子を付与するか選べます。

今回は中性条件での状態としたいので、上のように「pH = 7.0」としました。アミノ酸残基には酸性、中性、塩基性といろいろな官能基がありますが、設定したpH条件下の適切な状態にそれぞれ自動的に水素原子を付与してくれるようです。
つづいて同じ画面で水ボックスの追加を行います。Continueを押さずに次項をご覧ください。
4-6. 水ボックス(+イオン)の設定
最後に水ボックスの設定を行います。先の項で見た画面の下部「Add Water/Membrane」でAdd water boxにチェックを入れると下図のように設定画面が出てきます。

まず、水ボックスのサイズと形を設定します。今回は「タンパク質構造を取り囲むように水の層を追加(Padding)」します。
上図のようにSpecify a padding distanceを選択し、「Padding Distance (nm): 1.0」、「Box Shape:Cube」としました。これで厚さ1.0 nmの水の層で、全体が立方体(Cube)状になるようにタンパク質を囲む水ボックスができることになります。
Cube以外のボックスの形としてはTruncated octahedron、Rhombic dodecahedronといったものがあります。(境界条件に関係する設定項目のようですがどれがいいかよくわかりません。)
次に、イオンを追加します。イオン濃度(Ionic strength (molar))と、イオンの種類(Positive ion、Negative ion)を選びます。上図では0.15 Mの濃度でNaClを追加しています。他に利用可能なイオンはプルダウンの中身でご確認ください。
以上で前処理の設定は全て終わりです!Continueを押してください。
4-7. 処理後のPDBファイルの保存
PDBファイルの前処理が全て終わると、そのままシミュレーション条件の設定画面に遷移します。この記事ではこの先までは進みません。
下図のような画面になるのでSave Processed PDB File を押して前処理が終わった状態のPDBファイルを保存することができます。

3poz-processed.pdbという名前のファイルが保存されました。PyMolで描画すると以下のようでした。

前処理の途中で欠けている残基のうち鎖の途中の箇所は追加しましたがどのようになっているでしょうか?処理前後の構造を重ねて表示してみました。グリーンが処理前(3poz.pdb)、ブルーが処理後(3poz-processed.pdb)です。*4

上図の通り、処理後は残基が追加され実線で表示されています。PDBFixerの機能の一つに「欠けているループの構築」があげられていましたが確かに実行されていますね!
5. おわりに
以上、今回はMD計算を行う前のタンパク質の処理について試してみました。
OpenMMの提供するGUIツール OpenMM SetupのPDBファイル前処理機能を利用しました。指示に従ってボタンを押していくだけで使えてわかりやすいですね!
処理に必要な項目と操作の流れが大体わかったので、次回、同じ操作をPythonからPDBFixerを使って実行する方法を試したいと思います。
計算関係は前処理が大事とよく言われますが、具体的にどうすれば良いかわからないので難しいです。こればっかりは計算対象ごとの各論なので仕方ないのかもしれません。*5
今回も色々と間違いが多そうです。ご指摘いただければ幸いです。
ではでは!
MD計算のためのオープンソースソフトウェアをさらっと
前回、前々回の記事でMaking-it-rainというプロジェクトを利用して分子動力学(MD)計算をGoogleColab上で実行できることと、そのパラメータについてお勉強してみました。
計算科学の分野はオープンサイエンスの流れが進んでいて、MD計算に関連するオープンソースソフトウェア(OSS)も色々とあるようです。Making-it-rainもOSSをベースに構成されています。
せっかく遊ぶならどうやって動いているのかも知りたいよね!
というわけで(?)、Makig-it-rain(とついでにTeachOpenCADD)を参考に、MD計算に関するオープンソースツールにどんなものがあるか?さらっと眺めてみました!

1. 取り上げるツール
この記事ではMaking it rainとTeachOpenCADD (T19, T20)で出てくるツールをコピペしていきます。参考資料の都合上、ここでとりあげるツールはタンパク質(+リガンドの複合体)のMD計算に偏っていそうです。ご留意ください。
こんな感じのツールが出てきました。
- MD計算本体を実行するためのツール
- 分子力場(低分子用?)
- 計算対象の前処理やファイルの変換
- 結果(トラジェクトリ)の解析
いっぱいだ!
2. MD計算OSSの本命? OpenMM
2-1. 概要
まずはOpenMMです。これが本丸(?)のソフトみたいなので、最初からクライマックス。
有名なソフトらしく日本語で検索してもWikipediaの記事(OpenMM - Wikipedia)や、使い方を解説してくださっている記事がヒットします。
OpenMMは、分子動力学シミュレーションのためのオープンソースのライブラリであり、パンデ研究室によって管理されている。Python、C++、Fortranなどの言語に対応しており、GPUで高速化する機能を持っている。また、AMOEBA、GROMACS、AMBERなどの他の分子シミュレーションツールとのインタフェースも可能である。スタンフォード大学の研究者ビジェイ・S・パンデが作成したこのプロジェクトは、GitHubで自由に利用できるようになっている。(Wikipedia - OpenMMより引用)
こんな感じ。
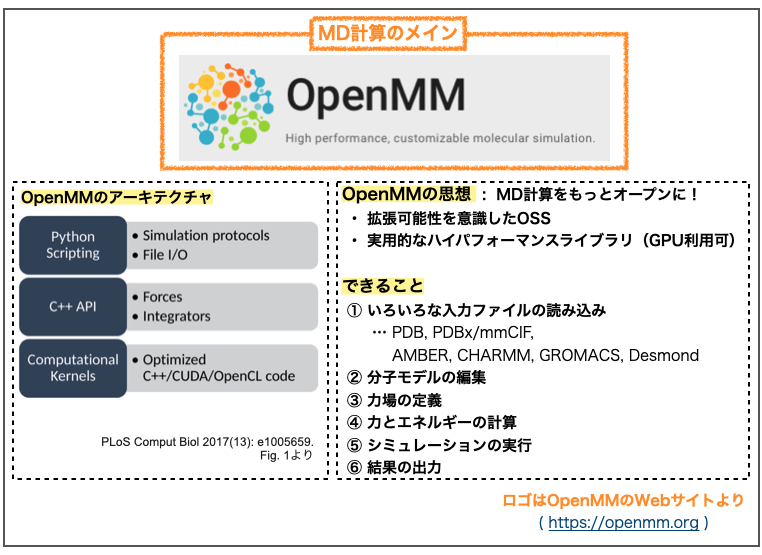
基本的にMD計算に必要なものが揃って提供されているようですね!ファイルの読み込み(①)から、シミュレーションの系の設定(②, ③)と計算の実行(④, ⑤)、結果の出力(⑥)までできるそうです。
2022年現在のバージョンはOpenMM 7.7で、2017年にversion 7の論文が出ています。
- Webサイト:OpenMM
- GitHub:openmm
- ドキュメント:User Guide
Webサイト上でチュートリアルや動画が公開されていたり、OpenMMを利用したソフトウェア・プロジェクトの紹介がされていたりと、ライブラリのエコシステムの維持・発展を意識した運営がされているのがとても魅力的です。
また、初学者にとって嬉しいことにシミュレーション条件をセットアップするためのGUIツールOpenMM Setupも提供されています。

英語 & CUI だと諦める私にとっては最高です!
2-2. OpenMMのインストール
condaがあればOpenMMのインストールは簡単です。(OpenMMユーザーガイド-Installing OpenMM)*1
conda install -c conda-forge openmm
GPUがある場合はインストール時にドライバをオプションで設定できるそうです。(私はMac Book Pro 2017でNvidia GPUもAMD GPUもないです。。。)
インストールがうまくいったかどうかは次のコマンドで確認できます。ターミナルにコピペ。
python -m openmm.testInstallation
こんなのでました。
OpenMM Version: 7.7 Git Revision: 130124a3f9277b054ec40927360a6ad20c8f5fa6 There are 3 Platforms available: 1 Reference - Successfully computed forces 2 CPU - Successfully computed forces 3 OpenCL - Successfully computed forces Median difference in forces between platforms: Reference vs. CPU: 6.3052e-06 Reference vs. OpenCL: 6.7302e-06 CPU vs. OpenCL: 9.02393e-07 All differences are within tolerance.
Successfullyっていってるからたぶん大丈夫。
2-3. OpenMM Setupのインストール
同様にOpenMM Setupもcondaでインストールできます。
conda install -c conda-forge openmm-setup
インストールできたら以下のコマンドでGUIが立ち上がります。
openmm-setup
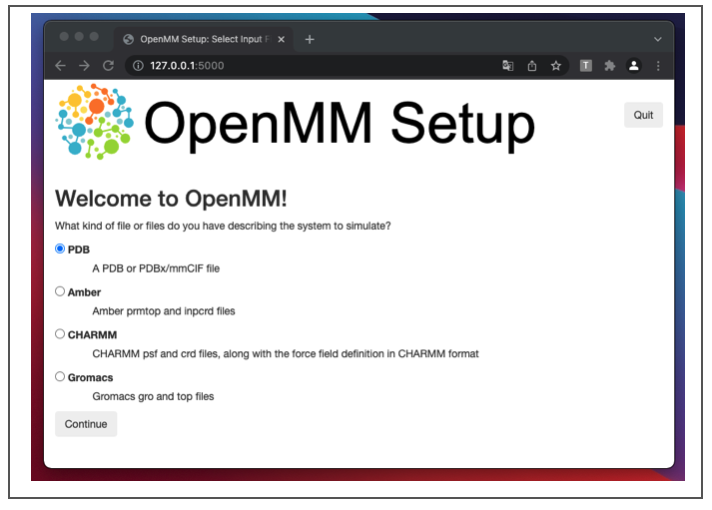
ブラウザで立ち上がりました!ターミナルには以下のような表示が出ました。
* Serving Flask app 'openmmsetup.openmmsetup' (lazy loading) * Environment: production WARNING: This is a development server. Do not use it in a production deployment. Use a production WSGI server instead. * Debug mode: off * Running on http://127.0.0.1:5000/ (Press CTRL+C to quit)
ブラウザが自動で開かなくても「http://127.0.0.1:5000/ 」をコピペしてアクセスすればOKで、終了したければ「CTRL + C」だそうです。Jupyter notebook使う時と一緒ですね。
このまま遊びたいところですが話が逸れるので次回にお預け。
3. 分子力場プロジェクト Open Force Field
3-1. 概要
つづいて分子力場のプロジェクトOpen Force Field(OpenFF)のご紹介です。
分子力場自体はOpenMMのツール群にも用意されていて、OpenMM ForceFields(GitHub)で利用することができます。こちらを利用することで、生体分子をメインにしたAmberやCHARMMといった分子力場や、低分子のためのGAFFやSMIRNOFFといった分子力場を使えるようになります。
なお、OpenMM ForceFieldsもOpenMM本体同様、condaでインストールできます。
conda install -c conda-forge openmmforcefields
で、OpenMM ForceFieldsで提供されている低分子の分子力場SMIRNOFFを作成したのがOpen Force Field イニシアチブです。
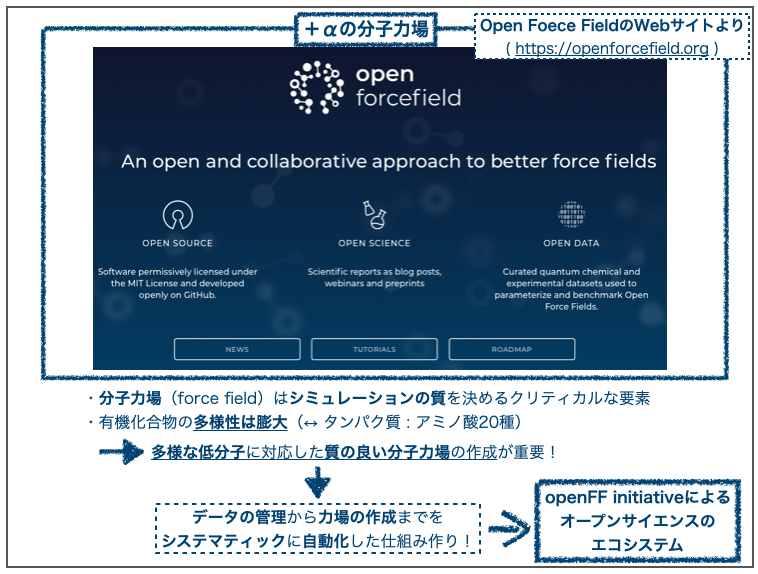
タンパク質の構成要素は凡そ20種に還元できますが、低分子は原子の種類・結合パターンがより多様で複雑です。低分子を対象にシミュレーションを行うには、広大なケミカルスペースに対応した精度の高い分子力場の作成が肝になります。
科学の発展(ex. 新規合成方法の開発)に伴ってケミカルスペースは継続的に発展しつづけるため、対応する分子力場も継続的に改善していく仕組み(エコシステム)作りが求められます。
「それならオープンサイエンスの出番でしょ!」
ってことで(?)、できたイニシアティブがOpen ForceField initiativeです。基になるデータ管理から力場の作成までをオープンかつ再現性よく行うため、システマティックに自動化した仕組みづくりが目指されています。

Open Force Field v.1.0.0(コードネーム Parsely)の論文は下記でプレプリントでもチェックできます。
DEVELOPMENT AND BENCHMARKING OF OPEN FORCE FIELD V1.0.0, THE PARSLEY SMALL MOLECULE FORCE FIELD Qiu, Y., Smith, D., Boothroyd, S., et al., J. Chem. Theory Comput. 2021, 17, 10, 6262–6280
chemRxiv DOI: https://doi.org/10.26434/chemrxiv-2021-l070l-v4 (CC BY 4.0)
Open Force Fieldで作成された力場は「SMIRKS-native Open Force Field(SMIRNOFF)」と名付けられています。SMIRNOFFのパラメータは量子力学(QM)計算で作成したデータセットに対してフィッティングされています。各パラメータの定義は、相互作用のタイプ(ex. 結合の伸縮)とSMIRKSパターンにそれぞれ基づいています。
SMIRKSは化学構造の線形表記の一種(SMILESとSMARTSのハイブリッド?)で、反応前後における分子の変化を一般化して記述できる表記方法のようです*2。論文のFigure 11を見るとイメージしやすいので引用しておきます。
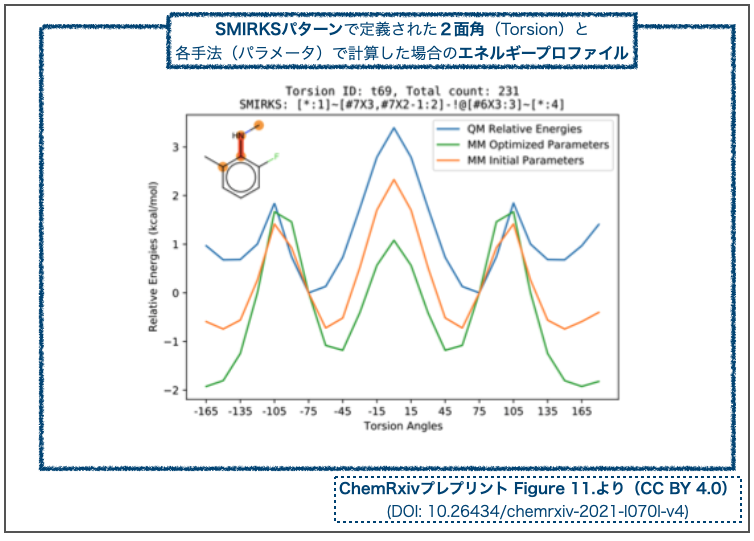
上図はグラフ左上の構造式でハイライトされた4原子の2面角の、角度に依存したエネルギー変化のプロットです。プロット上部の「SMIRKS」部分をみれば、パターンで部分構造を定義しているというのがわかりますね。
力場作成の詳細な方法や精度のベンチマークは論文をご参照ください。
3-2. インストール
Open Force Fieldを利用するにはOpen Force Field ToolkitをインストールすればOKです。condaがあれば簡単です。(openff toolkit - installation)
conda install -c conda-forge openff-toolkit
ドキュメントには使い方の実例がたくさん載っています(Examples using SMIRNOFF with the toolkit)。GitHubのレポジトリにリンクされていて、ノートブックやBinderでのデモも用意されているので要チェックだ!
…私はまだ一つも見れてません。すみません。
4. 準備ツール
つづいて、シミュレーションを行うための準備に関するツールです。モデル構造の修正(PDBFixer)や、ファイルフォーマットの変換(ParmED)といった操作に使えます。
4-1. PDBFixerでPDB構造を修正
タンパク質のシミュレーションを行う際、基になる構造情報をPDBから取得するケースが多いと思います。ですが、PDBに登録されている情報が必ずしもシミュレーションに適しているとはいえないため、目的に合わせた修正を行う必要が生じます。
PDBFixerはシミュレーションに合わせたPDBファイルの修正(fix)を行うためのツールで、OpenMMのエコシステムに組み込まれています。
こんな感じのことができます。

PDBFixerのインストールもcondaでOKです。
conda install -c conda-forge pdbfixer
PDBFixerはコマンドラインアプリケーションとしても使えますが、Python APIが用意されていてそちらの方がpowerfulに使えるそうです。
4-2. ParmEdでフォーマットを変換
計算で遊ぶときの一つのネックは、色々ソフトウェアがあって、それぞれに異なる拡張子のファイルを使っている場合です。MDシミュレーションにもAmberやCHARMMといった複数のソフトがあります。
ParmEdは異なるファイルフォーマット間で変換や読み書きができ、この問題を解決してくれます。生体分子シミュレーションの主要なソフトウェアをカバーしていて、Open Force Field Toolkitの使用例でも構築した系の変換に使われています(Export OpenFF-generated OpenMM System to AMBER and GROMACS files)。

- GitHub : ParmEd
- ドキュメント : ParmEd
- 文献:Shirts, M.R., Klein, C., Swails, J.M. et al. Lessons learned from comparing molecular dynamics engines on the SAMPL5 dataset. J Comput Aided Mol Des 31, 147–161 (2017). DOI: 10.1007/s10822-016-9977-1
ParmEdのインストールもcondaでOKです。
conda install -c conda-forge parmed
本記事で参考にしている資料では、Making it rainのProtein-Ligand Simulationsで、タンパク質-リガンド複合体のシミュレーション系を構築する際に利用されていました。
5. 結果(トラジェクトリ)の解析
OpenMMはMD計算の準備と実行、結果の出力までできますが、得られた結果(トラジェクトリ)の解析には、それに特化したソフトウェアがいくつかあるようです。参考資料の中にはPyTraj、MDTraj、MDAnalysisというツールがでてきました。・・・そもそもトラジェクトリ解析にどのような手法があるのかわかっていないのでツール間の違いがよく分かりませんでした。
5-1. PyTraj
PyTrajはAMBERのAmber Toolsに組み込まれているcpptrajというプログラムをPythonから扱えるようにしたフロントエンドパッケージです。MDでよく見るAMBERプロジェクトと関わりがある点が魅力的です。
- GitHub: pytraj
- ドキュメント:pytraj 2.0.2.dev0
インストールはcondaでできますが、チャンネルがambermdである点に注意です(pytraj - Installation)。
conda install -c ambermd pytraj
ドキュメントにチュートリアルをたくさん用意してくださっているので、こちらをみれば大体どんなことができるのか分かりそうです。

参考資料では、Making it rainのトラジェクトリ解析(RMSD計算、PCA解析 etc.)は基本的にPyTrajで実行されています。
また、他の解析パッケージとの互換も意識されてるようで、ドキュメントにはmdtrajやmdanalysisとのインターフェイスについてのチュートリアルがあります。
5-2. MDTraj
つづいてMDTrajです。こちらもPythonで利用可能なトラジェクトリ解析ツールです。OpenMMの開発も行っているStanford大学のPande Labによるプロジェクトという点で魅力的です*3。
- GitHub : mdtraj
- ドキュメント:MDTraj
- 文献:McGibbon, R.T., Beauchamp, K.A., Harrigan, M.P., et. al., MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories Biophysical Journal 2015(109)1528–1532 DOI: 10.1016/j.bpj.2015.08.015
MDTrajもcondaでインストールできます(MDTraj - Installation)。
conda install -c conda-forge mdtraj
こちらもドキュメントに使用例がコードとともに掲載されています。
参考資料では、TeachOpenCADDのT019 · Molecular dynamics simulationで使われていてます。トラジェクトリの解析ではなく、それぞれ別に用意したタンパク質とリガンドのモデルを一つの複合体としてシミュレーションするためにマージするツールとして使われています。
5-3. MDAnalysis
さいごにMDAnalysisです。これもPythonで利用可能なトラジェクトリ解析ライブラリです。ドキュメントだけでなくブログやビデオチュートリアルがあったりと、開発が活発に続けられている感じなのが素敵です。
- GitHub:MDAnalysis
- ドキュメント:MDAnalysis documentation
- 文献①:R. J. Gowers, M. Linke, J. Barnoud, T. J. E. Reddy, M. N. Melo, S. L. Seyler, D. L. Dotson, J. Domanski, S. Buchoux, I. M. Kenney, and O. Beckstein. MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations. In S. Benthall and S. Rostrup, editors, Proceedings of the 15th Python in Science Conference, pages 102-109, Austin, TX, 2016. SciPy. doi:10.25080/Majora-629e541a-00e
- 文献②:N. Michaud-Agrawal, E. J. Denning, T. B. Woolf, and O. Beckstein. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 32 (2011), 2319--2327. doi:10.1002/jcc.21787
MDAnalysisもcondaでインストールできます(MDAnalysis - Installation)。
conda install -c conda-forge mdanalysis
JupyterLabで使いたい場合は、合わせてnodejsをインストールして設定しておくと良いそうです(解析の際にプログレスバーが表示できたりするらしい?)。
conda install -c conda-forge nodejs jupyter labextension install @jupyter-widgets/jupyterlab-manager
使用例やチュートリアルはMDAnalysis - User Guideに詳しいです。
参考資料では、TeachOpenCADDのT020 · Analyzing molecular dynamics simulationsでトラジェクトリ解析に使われています。RMSDの計算やタンパク質-リガンド間の相互作用(原子間距離、水素結合)の解析が行われています。
6. おわりに
以上、今回はオープンソースソフトウェアを使ってMD計算を試す際に便利そうなツールをひろってみました。といってもMaking it rainとTeachOpenCADDで出てくるソフトを列挙しただけですが、、、
「実際にどう使うか?」までは踏み込めず、「(似たような)ツール間の違い」も分かりませんでしたが、とりあえずドキュメントのありかがわかった!!
複雑な計算科学の分野で、オープンサイエンスにむけた取り組みが進んでいるのはとてもありがたい話ですね。エコシステムとして維持・発展させるための取り組みには科学に限らず参考にすべき点が多い気がします。・・・まず、かわいいロゴが大事!!
オープンマインドにあふれた偉大な先人たちのおかげで、いろいろなドキュメントやチュートリアルが手に入るようになりました。・・・が、残念なことにレベルが低い私はノートブックの最初に列挙されているimport ~~ as ~~が追いきれず、途中で「これなんのライブラリ使ってるんだ??どっから来た?」「自分で遊ぶときには何をインストールして使えばいいんだ???」 となってしまいます。
今回、なんとなくMD計算で利用できそうなソフトの名前がわかったのでヨシ!
他にもおすすめのソフトウェアがあったら教えていただけると嬉しいです。間違いが多そうなのでご指摘いただければ幸いです。
ではでは。
Google CouldでMD計算をお試し!ーMaking it rainー
タンパク質がぐにゃぐにゃ動く分子動力学(MD)計算かわいいですよね。下はD.E. Shaw Research("DESRES")のツイートで、彼らの開発したスパコン(アントン - wikipedia)で計算されたものらしいです*1。
50 2-µs simulations of FDA approved or investigational drug molecules binding to a construct of the #SARSCoV2 trimeric spike protein. Full data at https://t.co/CQVDq1clZB pic.twitter.com/LEQ9RqBKLy
— D. E. Shaw Research (@DEShawResearch) 2020年5月6日
「お高いパソコンがないとMDで遊べないのかな?」と思ってましたが、Twitterで「低分子ならGoogle Colabでも結構できるよ!」「Making it rainが参考になるよ!」って教えていただきました。@ylogtさん、ありがとうございました!
というわけで、とりあえずMakig-it-rainをそのまま実行した結果のご報告です。*2

1. Making it rain?
1-1. 概要
Making-it-rainは計算リソースが必要な分子動力学(MD)計算を、誰でもお手軽にできるようにしよう!というプロジェクトです。GoogleアカウントがあればOK!
Maiking-it-rainではOpenMMというオープンソースのMDシミュレーションライブラリをGoogle Colab上で実行することができます。ノートブックに従って順番に実行することで、①MD計算の準備、②実行、③結果の解析まで行えます。また、Google Driveとの連携されているので、計算結果やノートブックの保存も容易です。
論文は以下。
- P.R. Arantes, M.D. Polêto, C. Pedebos, and R. Ligabue-Braun Making it Rain: Cloud-Based Molecular Simulations for Everyone
J. Chem. Inf. Model. 2021, 61, 10, 4852–4856
GitHubレポジトリは以下。
MD計算をハンズオンで学習するための教育や、資金の少ない研究室でもマイクロ秒スケールのシミュレーションができるようにするため開発されたそうです。ありがたいですね!
1-2. 論文の紹介(Zenodo公開版)
査読済みの論文はオープンアクセスではありませんが、プレプリントをChemRxivやZenodoで公開してくださっています*3。計算の中身やコードの詳細というよりも、プロジェクトのモチベーションや大枠の構成の説明と、実行例の紹介といった内容になっています。
Making it rainの基本的構成は、① 計算パワーの必要なMD計算をGoogle ColabのGPUで実行し、② 入手力ファイルや設定ファイル、結果といったデータ保存・管理をGoogle Driveで行うものとなっています。
Google Colab、Google DriveともにGoogleアカウントがあれば無料で利用できますが、有償版ではより長時間・高容量の利用ができます。*4

計算時間の目安として、リゾチーム(lysozyme)を題材に1μsのシミュレーションを行った例が論文に書かれています。より高性能のGPUが使える有償版(Colab Pro)では、平均して約230ns/dayのシミュレーションが可能で、1マイクロ秒には6日あればOKだったそうです。
Colabには時間制限(有償版でも1日)があるため、Making it rainは長時間の計算を分割して実行できるように作られています。論文の例では、1マイクロ秒全体のトラジェクトリを20n秒ごと、50個のストライドに分割しています。各ストライドの終わりに、その時点での系の状態を保存したファイル(.xml)をOpenMMが出力してくれるので、出力ファイルを元に再度計算を実行することで連続したシミュレーションができる、とのことです。例では5回実行することで計1μsの結果を得ています。
1-3. ノートブックの種類
Making it rainではGoogle Colabで実行可能なノートブックが複数用意されており、それぞれ計算内容や計算方法が異なっているようです。論文では3つですが、現在はさらに3つアップデートされ、計6つのノートブックが利用可能です。
いずれもGitHubレポジトリのトップ(README)から利用可能で、こんな感じ。
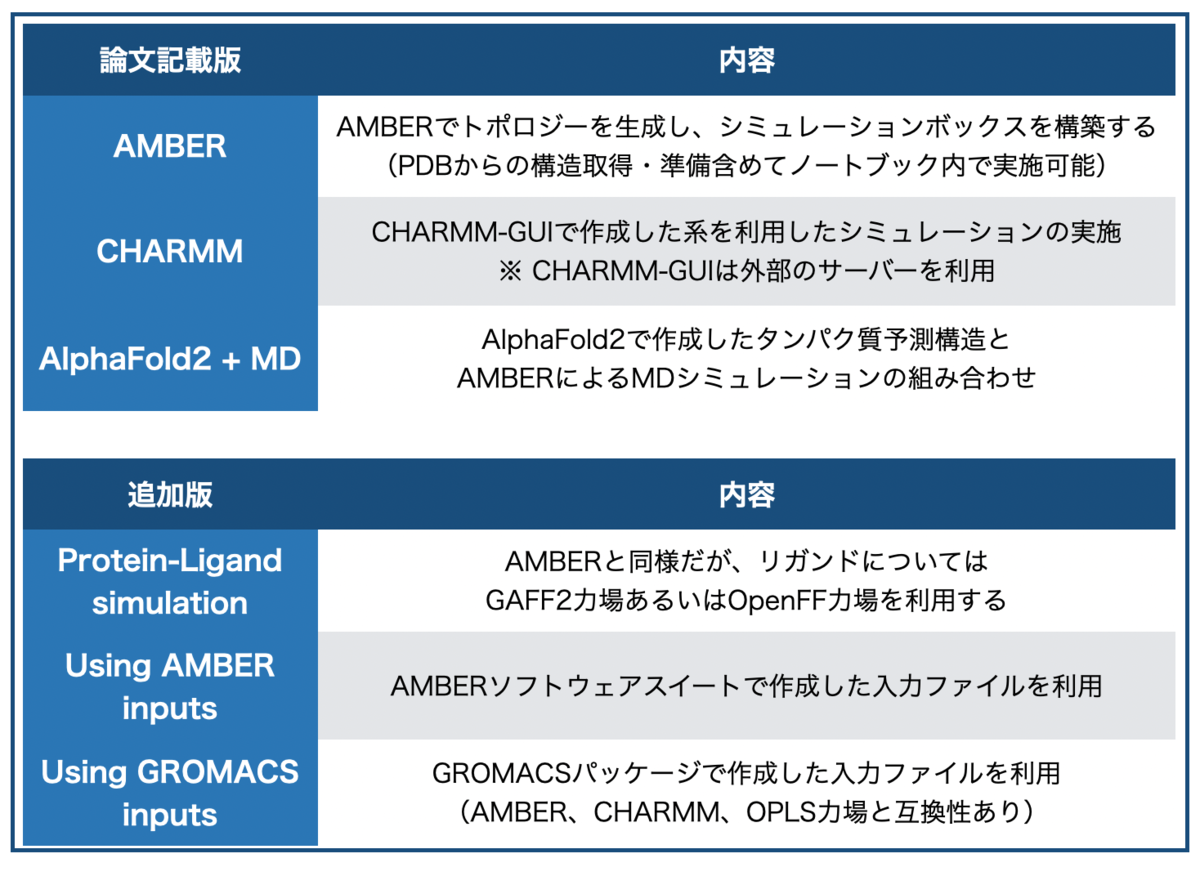
入力ファイルを作成するソフトウェアや利用する力場によってノートブックが分かれているようですが、MD計算を勉強したことがないので全く違いがわかりません。すみません。
よくわからないけどタンパク質モデリング(AlphaFold2+MD)やリガンドとの相互作用(Protein-ligand simulation)といったトピックもおさえてあるのが魅力的!!
以上、Making it rainの概要でした。
2. 使ってみた
2-1. AMBERノートブックの流れ
ざっとMaking it rainの目的が把握できたので早速遊んでみましょう!今回はノートブックのうち「AMBER」をそのままデフォルトで実行してみました。*5
AMBERノートブックの流れは以下のような感じでした。

「ノートブックを順番に操作するとどうなるか?」ビデオ形式のデモまで著者のPablo Arantesさんが用意してくださっています。ノートブックを開くとすぐに見られる(2分ちょっと)ので、流れを把握するのに便利です。親切!!*6
2-2. やってみた 〜MD計算の実行〜
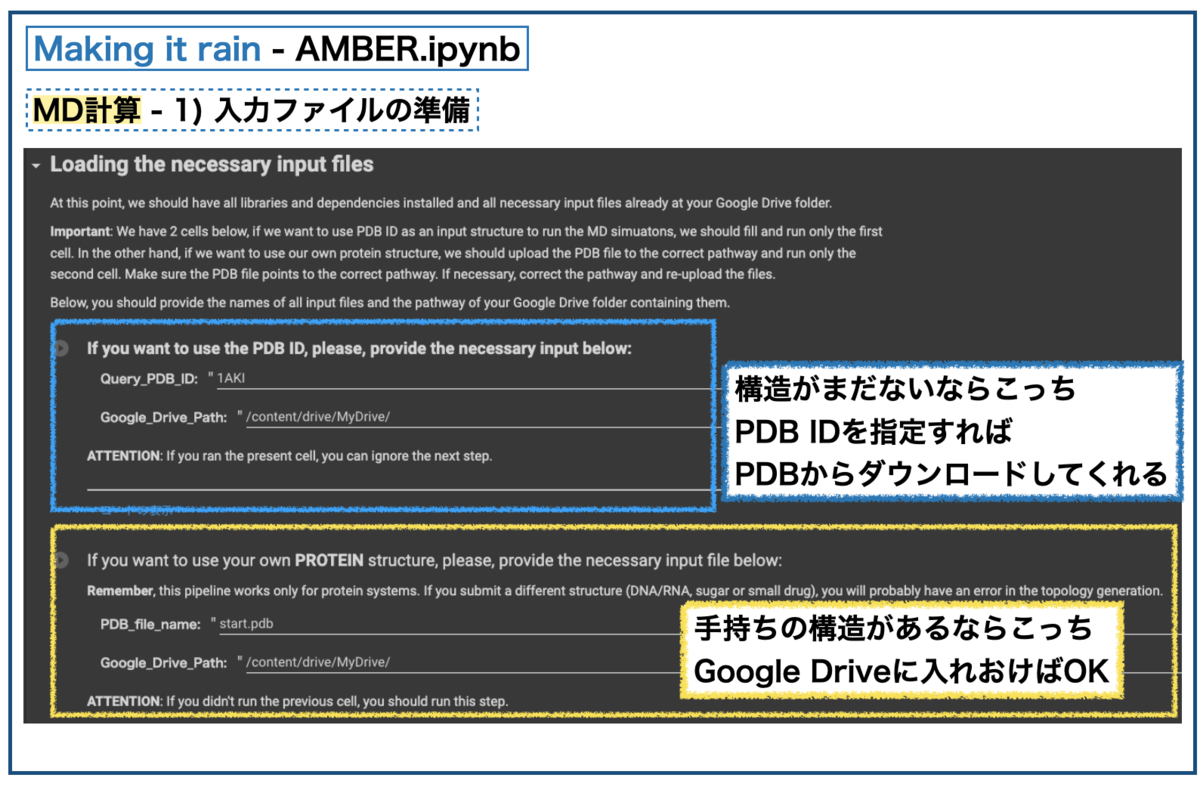
「AMBERノートブック」のデフォルトの題材は「ニワトリ卵白由来のリゾチームの水中における挙動」のシミュレーションでした。MD計算実行部分がどんな感じだったかご紹介します。
まず、初期構造の準備です。PDBからPDB ID: 1AKIを取得しています。手持ちの構造が使いたければGoogle Driveに入れておけば参照できます。
次にシミュレーションの系の設定です。水で満たされたボックスにタンパク質が一つ配置されます。さらにイオン(NaCl)を加えて系を中和しておきます。
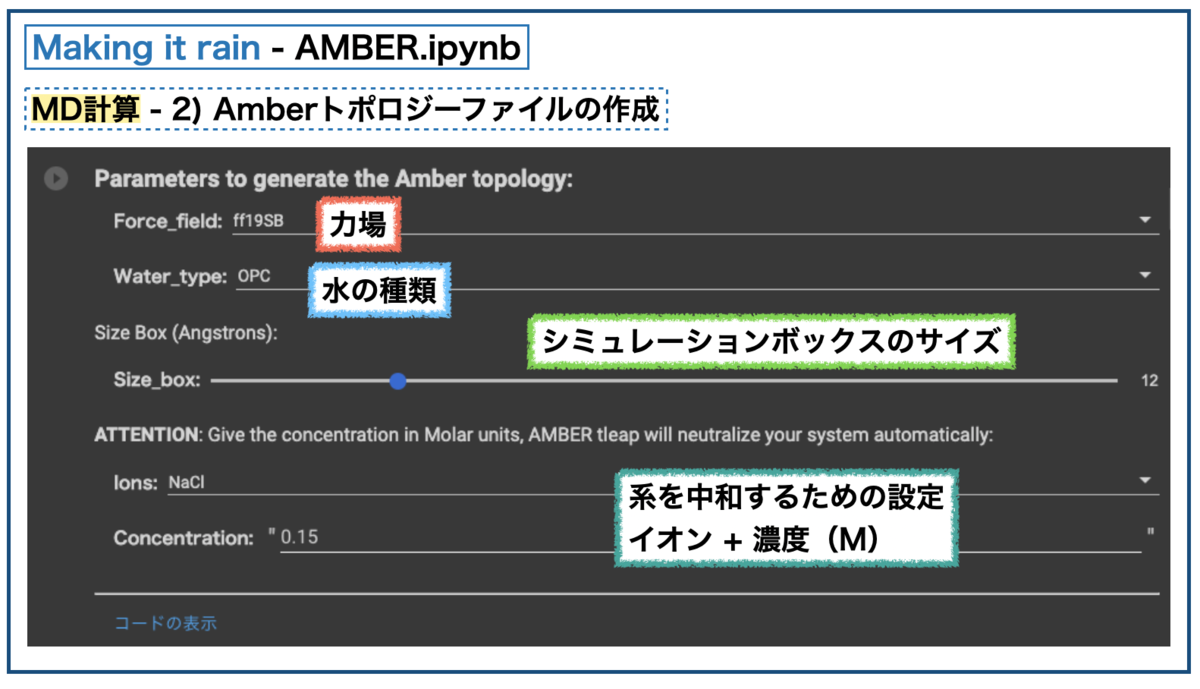
上がトポロジーファイルのパラメータ設定です。力場(force field。上ではff19SB)や水の種類、ボックスサイズ、イオンといった項目があります。これを元にMDシミュレーションのための系のパラメータがつくられます。
@Ag_smith先生の以下の記事に詳しいですが、AmberではtleapというモジュールでPDBファイルからトポロジーファイルを作成するそうです。実際にMaking it rainのコードを表示するとtleapを呼んでいます。
トポロジーファイル作成後、「シミュレーションの系がどのようにできたか?」、ノートブック上で3Dで可視化して確認できます。MD計算は時間が長いのできちんと準備できているか心配になりそうですが、ビジュアルで確認できると安心できていいですね!

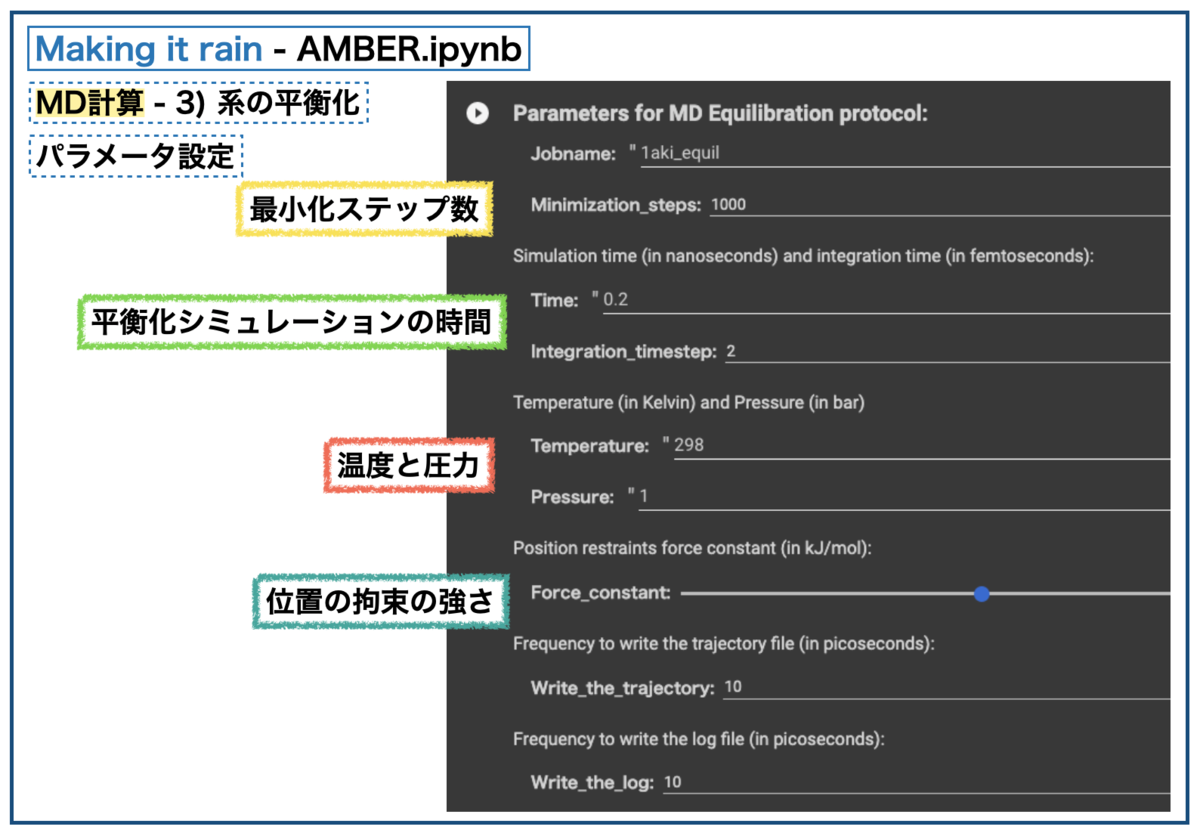
系が準備できたら平衡化(equilibration)を行います。MD計算ではシミュレーションの間、系の温度や圧力が一定の値を保つように計算を行うそうです。そのためにシミュレーションの本番前に短い計算で系をならしておくようです。*7
設定はこんな感じ。
先の@Ag_smith先生の記事から関連する解説を引用させていただきます。*8
ここからは水溶液中に存在するタンパク質を物理演算で動かしていくことになります。しかし、これまでの操作で作り出した初期座標では水溶液の様子を再現しているとは言えません。・・・(中略)・・・そこで、まずは系のエネルギーをある程度小さくしてから(=エネルギー最小化)、各原子に(温度に対するBoltzmann-distributionに従う範囲で)ランダムに速度を与え、徐々に系全体を設定温度まで引き上げていく(=平衡化)という予備動作を行うことで水溶液の状態に近づけていくという操作を行います。
①エネルギー最小化、②平衡化の順に実施するそうです。確かに、設定の最初にminimizationの文字が見えますね。
設定が終わったら平衡化シミュレーションの計算を実行します。NPTアンサンブルは物質の量(N)、圧力(P)、温度(T)が一定になるようにシミュレーションを行うそうです。「気温と大気圧に開放されているフラスコを用いた実験室条件に最も密接に対応している」らしい。。。(Wikipedia - 分子動力学法)

平衡化の長さは0.2nsとしていて、平均して約45ns/day(〜 0.03 ns/min)のスピードでシミュレーションできたようなので、計算時間は6分程度でした。"Time Remaining" で残り時間の目安がわかるのが親切ですね。
最後にシミュレーション本番(Production)を実行します。シミュレーションの長さはNumber_of_stridesとStride_Timeで指定し、2つの掛け算が得られる長さとなります。
例)100n秒シミュレーション = (ストライド数 10) x(各ストライドの長さ 10n秒)
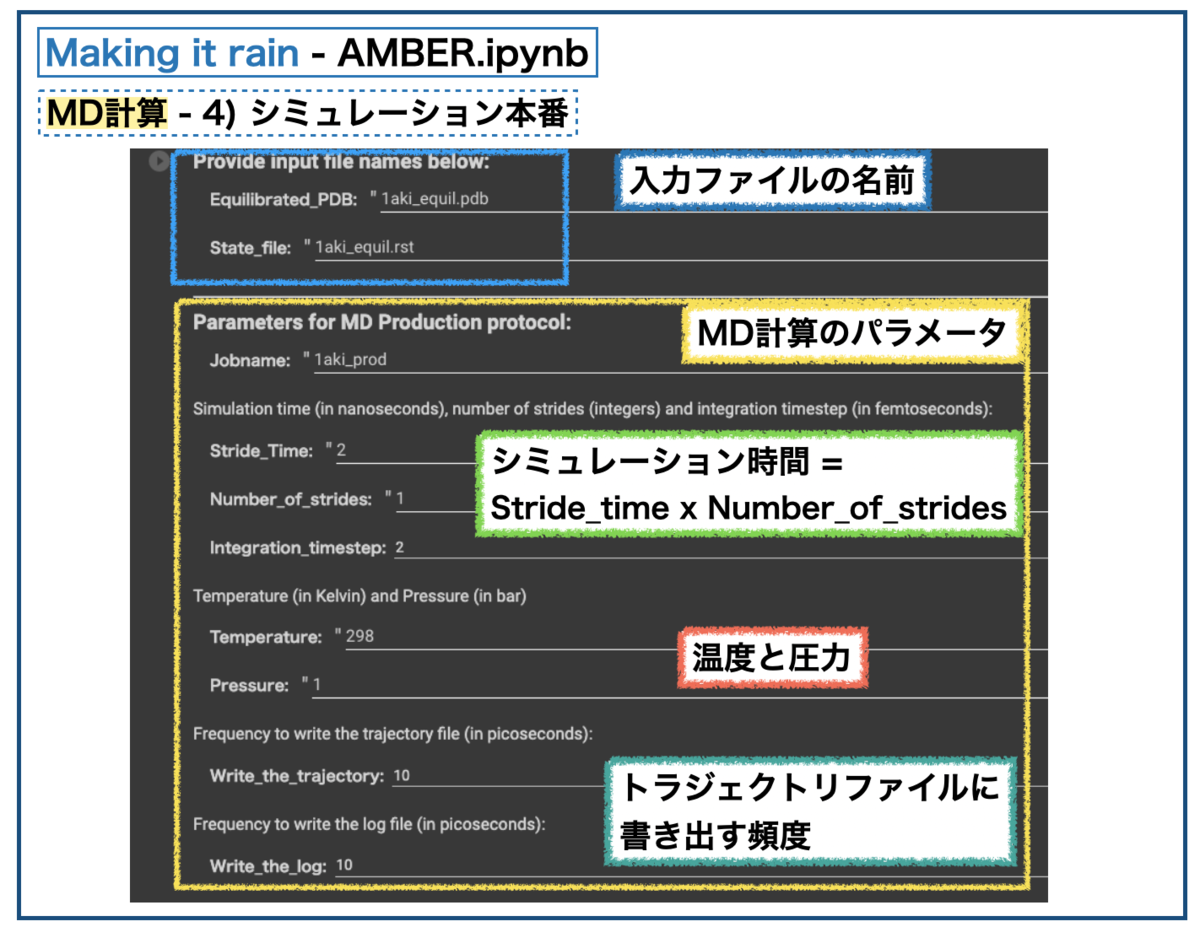
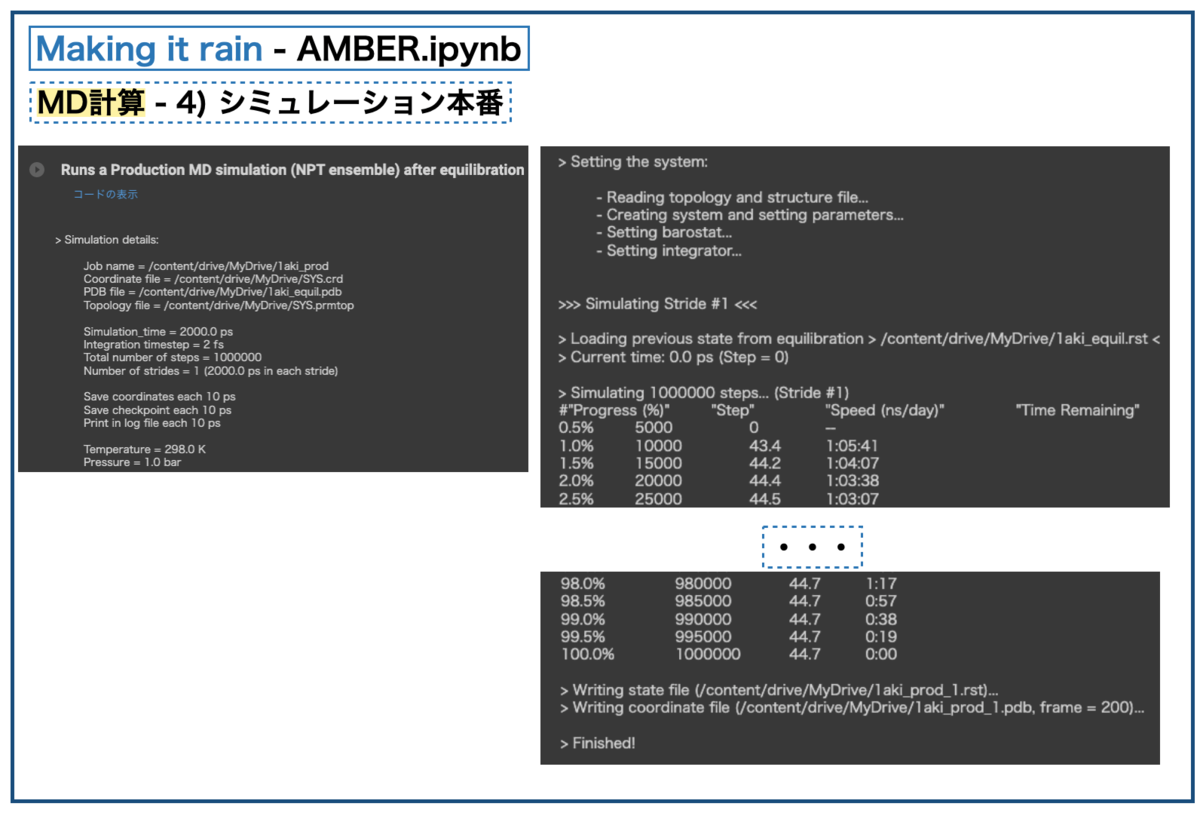
今回は全てデフォルトのまま2nsのシミュレーションを行いました。以下を実行し、計算時間は1時間ちょっとという感じでした。平衡化の10倍の長さなので、だいたい同じ感じのスピードで計算できた感じです。
MD計算が終わったら、シミュレーションの最後にトラジェクトリ(trajectory)ファイルを一つにまとめる操作を行います。
系に含まれる各原子の3次元座標の時間変化のことをトラジェクトリー(trajectory、軌跡)と呼ぶそうです*9。パラメータ設定で「トラジェクトリファイルに書き出す頻度」を10psとしたので、2nsのシミュレーションでは200個のファイルができることになります。これを一つのファイルにまとめます。
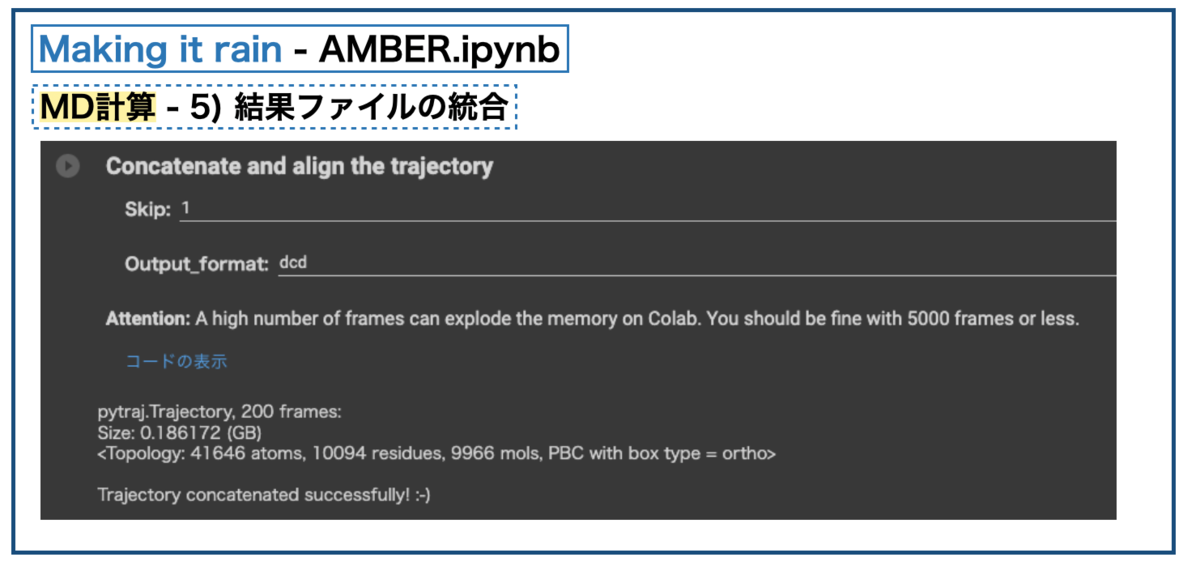
まとめると0.2GB弱でした。2nsで0.2GBなので1μsだと100GBくらいになりそうです。・・・でっかい。
2-3. やってみた 〜結果の解析〜
計算の実行とファイルの統合が終わったので、結果の解析部分を見てみましょう!
Making it rainのノートブックに用意されているので、引き続き実行結果をみていきます。
まずはトラジェクトリファイルを動画で見ます。「Load, view and check the trajectory」というセルを実行するだけ!
こんな感じ(以下はキャプチャのGIFです)

ピクピクしてる!!かわいい!
「動画もいいけど、もう少し定量的に結果を見たいよね!」というわけで、以下のような解析が用意されています。それぞれセルを実行するだけなので結果のプロットを貼っていきます。
まずは各アミノ酸残基のCα原子(中心炭素原子)についてこんな解析ができます。

①RMSD(根平均二乗変位, Root Mean Square Deviation)、②radius of gyration(慣性半径)、③RMSF(根平均二乗揺らぎ, Root Mean Square Fluctuation)です。①と②は時間に依存した変化と分布、③は残基ごとの値についてそれぞれプロットされています。
まったくどう解釈していいかわからない!!
①RMSDが0.5nsくらいまで増加傾向にあるのをみると、production前の平衡化が足りていなかったのでしょうか?
③RMSFでは末端以外にも残基40~50や70前後で少し大きくなっていますね。動画(GIF)で左側青色のループ部分に相当しそうなので、動きが大きそうな部位を数値化できてる感じでしょうか??。
残りの解析結果も貼っておきます。こんな感じ。

2D RMSD、PCA、Pearson's Cross Correlation (CC, ピアソン相関)。。。よくわかりませんでした。
結果の解析部分は以上です。
3. おわりに
以上、今回はGoogle Colab上でMD計算を実行できるMaking it rainと、実際に実行してみた結果のご紹介でした。
Making it rainではオープンソースのMD計算ソフトopenMMを利用しています。こちらはTeachOpenCADDでも使われているそうです。@iwatobipen先生が記事「Make mdtools for openmm」で紹介してくださっていました。*10
専門外の分野でも、オープンソースのツールがあったり使い方の解説があると「ちょっと遊んでみようかなー」となるので嬉しいですね!Making it rainはさらに低コストで高コストなMD計算を実施するフレームワークを提供してくれていて、とてもありがたいです。
残念ながら専門外すぎて、どういう設定で計算して、結果をどう評価していいのか全くわかりませんでした。不勉強ですみません。。。
ところで「Make it rain」という単語、直訳すると「雨を降らせる」ですが、Googleで検索すると強面のお兄さんたちと紙幣が舞う画像がでてきます。スラングで「大金をばら撒く、札束をまき散らす」といった意味があるとか。。。
クラウド(雲)コンピューティングから高価なデータの雨を降らせよう(たくさんお金がないとできないような計算をいっぱいやろう)なんて、意味が込められてたら面白いですね。
今回も色々と間違いが多そうです。ご指摘いただけると嬉しいです。
ではでは
追記:日本語化ノートブック
Making it rainのノートブックを日本語化したものをGitHubに入れておきました。ほとんどDeepL翻訳そのままで、動作確認もまだできていませんがご参考までにどうぞ。一応Colabでも動かせるはずです。
*1:引用したツイートと関連するMD計算の結果はDESRESのHPで公開されています(CC-4.0)。Molecular Dynamics Simulations Related to SARS-CoV-2。
*2:下の図はGitHubのFigureといらすとやさんのイラストを利用させていただいています。
*3:ChemRxiv上のライセンスはCC BY NC ND 4.0です。ZenodoはCC BY 4.0になっていますがサイトの設定か論文本文かどちらかわからなかったです。この記事ではZenodo公開版を参照させていただいています
*4:無償版/有償版の費用や性能は変わる可能性があるので適宜検索してください。Google Driveの有償版としてはGoogle Oneの情報を記載しました。
*5:何もわからないので一番上にあるやつをクリックしただけ。。。
*6:デモ動画はYouTubeにありますが「限定公開」設定なのでここにはリンクを貼らないでおきます。ノートブックをひらけばすぐわかります。
*7:全然違う話題ですけど、平衡化を最初に行うのベイズ推定のマルコフ連鎖モンテカルロ法(MCMC)使ったパラメータ推定の計算みたいですね。
*8:ただし引用記事はこの部分でAmberではなくGROMACSというソフトに切り替わっています
*9:参考:古明地 勇人, 上林 正巳, 長嶋 雲兵 生体分子の分子動力学シミュレーション(1)方法 J. Chem. Software, 2000(6)1–36
*10:TeachOpenCADD、インストールで力尽きて新しいトークトリアルの中身をまだ見られていないです。すみません。
2Dも3Dも!Panel-Chemistryでまとめて描画しよう!
前回の記事でJupyter notebookで構造式エディタを使う方法を調べました。
もっといい方法がありました!その名もPanel-Chemistry!!
2Dの構造式(JSME Editor)だけでなく、3Dの描画(NGL Viewer, Py3DMol)もカバーしてくれています。こりゃ便利!
1. Panel-Chemistry?
とりあえずデモが格好いいですよ!*1
Panel-Chemistryは、化学分野の探索的データ解析・ビジュアリゼーションをサポートするためのプロジェクトです。Pythonでデータ可視化するためのオープンソースライブラリであるHoloViz - Panelに化学ドメインを組み込んで構築されているのでPanel-Chemistryと名付けられているみたいです。
Panel-Chemistryがサポートしている可視化ツールは3つです。
- JSME Editor (化学構造式描画&編集、2D)
- NGL Viewer (タンパク質、DNA/RNA etc.の描画、3D)
- py3DMol (タンパク質、低分子化合物の描画、3D)
それぞれ対応可能なファイルフォーマットが異なります。2D、3Dの生物学・化学描画をカバーしてくれているのはありがたいですね!
# pipの場合 pip install panel-chemistry # condaの場合 conda install -c conda-forge panel-chemistry # もちろんmambaでもOK mamba install -c conda-forge panel-chemistry
とりあえず使用感を知りたいと言う方は、GitHubレポジトリのREADMEにBinderへのリンクが用意されているのでそちらをご利用ください。
2. 使ってみよう!
GitHubの例(examples)ほぼそのままで恐縮ですが、どんな感じに使えるかご紹介します*2。
サポートする可視化ツールそれぞれについて、①Jupyter notebook上で描画、あるいは②サーバーモードでアプリケーションとして利用する方法の2つの使い道があります。
2-1. JSMEの場合
2-1-1. Jupyter notebook上で使う
まずはJSMEをJupyter notebookで使う場合です。JSMEについては以下をご参照ください。
- Bienfait and P. Ertl, JSME: a free molecule editor in JavaScript, J. Cheminformatics 5:24 (2013)
panelとpanel_chemistryから使いたいツール(今回はJSMEEditor)をインポートしておきます。
import panel as pn from panel_chemistry.widgets import JSMEEditor
次にpanelのextension()でjsmeを読み込みます*3。以下のpn.extension()実行前に、上記の通りpabel_chemistryを読み込んでおく必要があることに注意です。
pn.extension("jsme", sizing_mode="stretch_width")
panel可視化スペースのレイアウトはcolumn()を使って設定できます。JSMEEditorを以下のようにレイアウトに追加するだけでもう使えます。
editor = JSMEEditor(height=500)
pn.Column(editor)

JSMEが起動しました!簡単!
さらに便利な機能は、JSME起動時に同時に指定した構造式が描画された状態で起動できることです。例えばSMILESで構造を指定するなら以下のようにすればOKです。
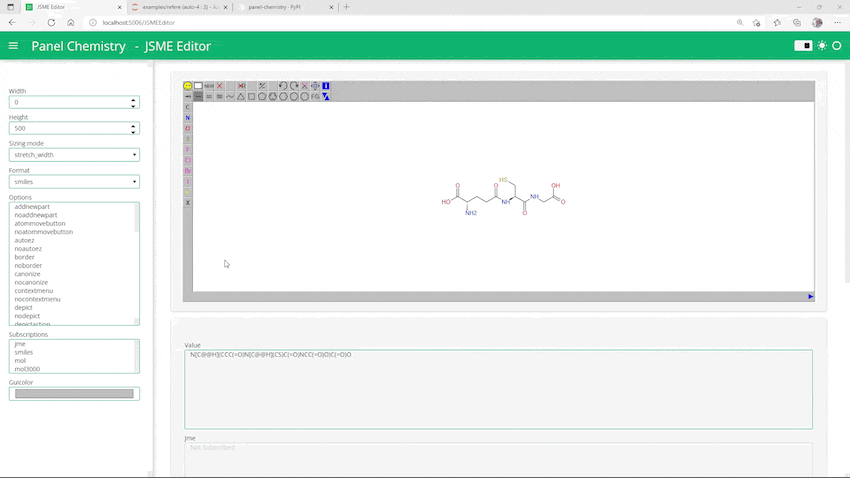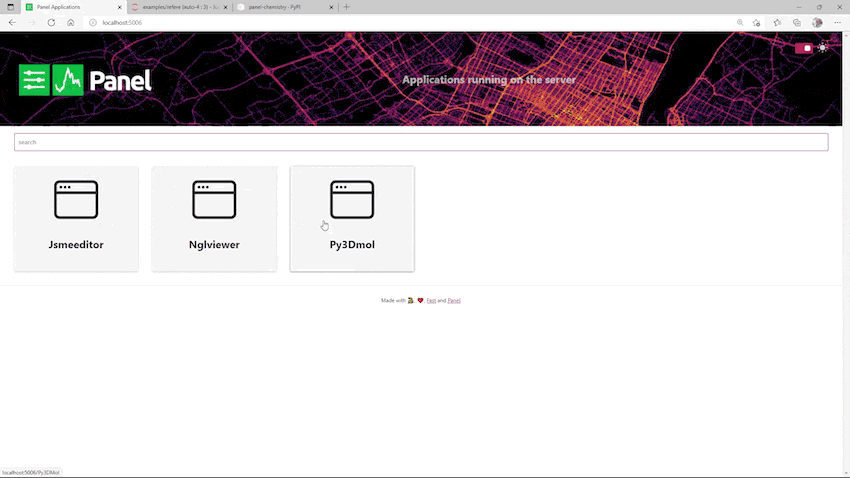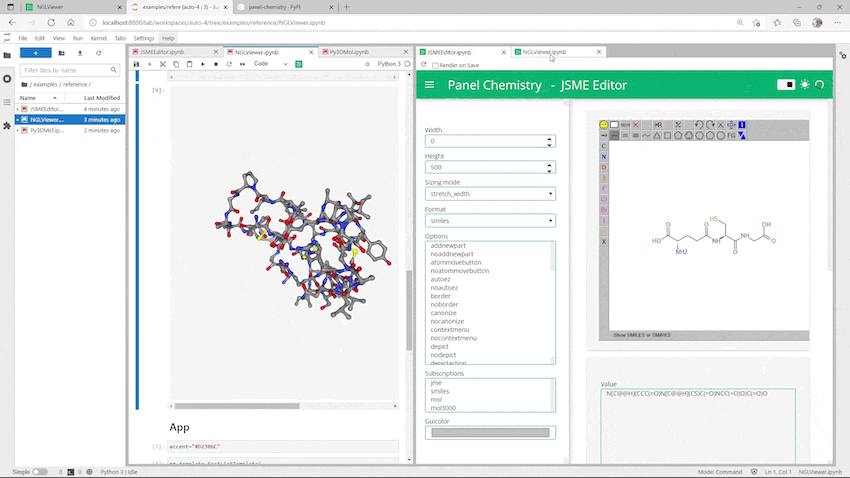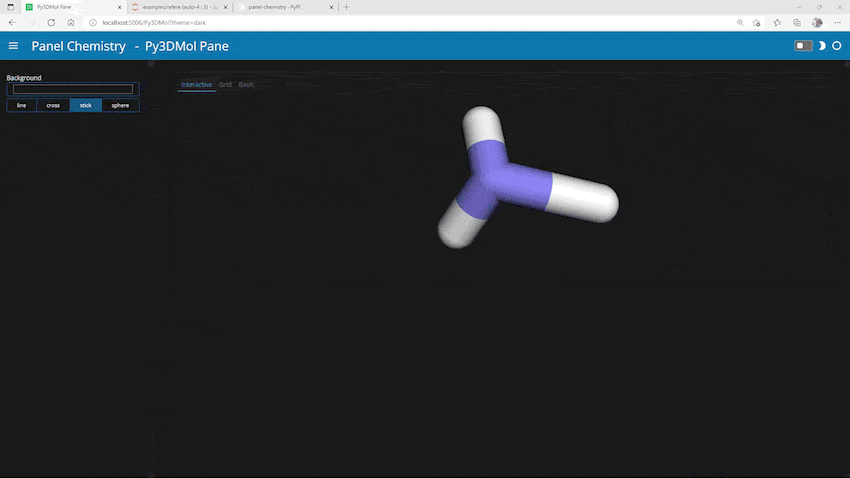
smiles="N[C@@H](CCC(=O)N[C@@H](CS)C(=O)NCC(=O)O)C(=O)O" editor = JSMEEditor(value=smiles, height=500, format="smiles") pn.Column(editor, editor.param.value)

上図の通りグルタチオンが描かれた状態でJSMEが起動しました。またpn.Column()でeditorに加えてeditor.param.valueもレイアウトに追加しているので、上図下部のようにvalueに格納されたSMILESも表示されています。
valueは他にもmolやsdf形式も使えます。例えば以下のようにRDKitで作成したMol形式の座標を読み込むこともできます。
from rdkit import Chem toluene = Chem.MolFromSmiles("Cc1ccccc1") toluene_mb = Chem.MolToMolBlock(toluene) print(toluene_mb) """ RDKit 2D 7 7 0 0 0 0 0 0 0 0999 V2000 3.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 1.5000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 0.7500 -1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -0.7500 -1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -1.5000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -0.7500 1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 0.7500 1.2990 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 1 2 1 0 2 3 2 0 3 4 1 0 4 5 2 0 5 6 1 0 6 7 2 0 7 2 1 0 M END """
# mol形式の座標を読み込んで起動する editor = JSMEEditor(value=toluene_mb, height=500, format="mol") pn.Column(editor)

トルエンのように単純な構造だと有り難みは少ないですが、より複雑な分子を描画する時、SMILESから構築するとツールに依存した描画スタイルになってしまいます。複雑な分子を好みの向き・スタイルで描きやすいと言う点で、座標情報のあるMol形式が使えるのは良いですね!
2-1-2. アプリケーションとして使う
つづいてアプリケーションとして使う場合です。ビューのレイアウトといった諸々の設定をJupyter notebook上に書き込んだあと、コマンドラインでpanel serveコマンドを使うことでアプリケーションとして起動することができます。
この方法ではPanelのFastLstTemplateテンプレートを利用してレイアウトを作成します。最後に.servabl()でアノテーションをつけておくことでコマンドラインから起動できるようになります。
Panelのアプリケーションサーバーの仕組みはドキュメントのServer Deploymentをご参照ください。デフォルトのポートは5006なので、起動後http://localhost:5006にブラウザでアクセスすればアプリを利用できます。
設定例はこんな感じ。
# アプリケーションの見た目の色調設定 accent="#00A170" # メイン描画部位の設定 values = pn.Param(editor, parameters=["value", "jme", "smiles", "mol", "mol3000", "sdf"], widgets={ "value": {"type": pn.widgets.TextAreaInput, "height": 200}, "jme": {"type": pn.widgets.TextAreaInput, "height": 200}, "smile": {"type": pn.widgets.TextAreaInput, "height": 200}, "mol": {"type": pn.widgets.TextAreaInput, "height": 200}, "mol3000": {"type": pn.widgets.TextAreaInput, "height": 200}, "sdf": {"type": pn.widgets.TextAreaInput, "height": 200}, }) # サイドバーの設定 settings = pn.Param(editor, parameters=["height", "width", "sizing_mode", "subscriptions", "format", "options", "guicolor"], widgets={ "options": {"height": 300}, }) # テンプレートを利用したレイアウトの作成 + servable() pn.template.FastListTemplate( site="Panel Chemistry", title="JSME Editor", sidebar=[settings], main=[editor, values], accent_base_color=accent, header_background=accent, ).servable();
上記設定が完了したら、コンソール(私はmacなのでターミナル)でpanel serve ノートブック名を打ち込めばサーバーアプリケーションとして起動します*4。「PanelChemistry_JSME.ipynb」というノートブックならこんな感じ。
panel serve PanelChemistry_JSME.ipynb
もちろんJupyter notebook上で最初に「!」をつけてコマンドを実行してもOKです*5。
実行後「サーバーを起動したぜ!」「アプリケーションはここだぜ!( Bokeh app running at: http://localhost:5006/PanelChemistry_JSME )」みたいなことを言ってくるのでこのURLをブラウザにコピペすればアプリが使えます。
こんな感じ!

緑を基調にしてるのが素敵ですね!
JSMEを使う場合は以上です。
2-2. NGL viewerの場合
2-2-1. Jupyter notebook上で使う
つぎにNGL viewerの場合です。NGL Viewerについては以下をご参照ください。
AS Rose, AR Bradley, Y Valasatava, JM Duarte, A Prlić and PW Rose. Web-based molecular graphics for large complexes. ACM Proceedings of the 21st International Conference on Web3D Technology (Web3D '16): 185-186, 2016. doi:10.1145/2945292.2945324.
AS Rose and PW Hildebrand. NGL Viewer: a web application for molecular visualization. Nucl Acids Res (1 July 2015) 43 (W1): W576-W579 first published online April 29, 2015. doi:10.1093/nar/gkv402.
やることは大体一緒です。まずはライブラリのインポートとngl_viewerのpanelへの読み込みです。
import panel as pn from panel_chemistry.pane import NGLViewer from panel_chemistry.pane.ngl_viewer import EXTENSIONS
pn.extension("ngl_viewer", sizing_mode="stretch_width")
次いで、ビューワーの設定です。
描画する目的の構造はobjectパラメーターで設定し、①url like、②non-url like、③blobの3種類から選べます。①と②はPDBに対応するもので、①なら「'rcsb://1NKT'」、②なら「'1NKT'」のようにします。②はPDB IDだけで、自動的に「rcsb://」を保管してURLにして構造を持ってきてくれます。
③blobの時は合わせて拡張子(extension)パラメーターを設定する必要があります。pdbやcif等色々と使えるのでGitHubなどをご参照ください。
今回はGitHubのexample通りやってもRCSBからPDBファイルを持ってきてくれなかったので、別途ダウンロードした「1nkt.pdb」を使っています*6。
# ビューワーと読み込む構造の設定 viewer = NGLViewer(object="1nkt.pdb",background="#F7F7F7", min_height=700, sizing_mode="stretch_both") # パラメーター設定 settings = pn.Param( viewer, parameters=["object","extension","representation","color_scheme","custom_color_scheme","effect",], name="⚙️ Settings" ) # 入力ファイルの設定 file_input = pn.widgets.FileInput(accept=','.join('.' + s for s in EXTENSIONS[1:])) def filename_callback(target, event): target.extension = event.new.split('.')[1] def value_callback(target, event): target.object = event.new.decode('utf-8') file_input.link(viewer, callbacks={'value': value_callback, 'filename': filename_callback}) # レイアウトの設定 header = pn.widgets.StaticText(value='<b>{0}</b>'.format("💾 File Input")) file_column = pn.layout.Column(header, file_input) layout = pn.Param( viewer, parameters=["sizing_mode", "width", "height", "background"], name="📐 Layout" )
pn.Row(
viewer,
pn.WidgetBox(settings, layout, width=300, sizing_mode="fixed",),
)

できました!
2-2-2. アプリケーションとして使う
つづいてアプリケーションとして使う場合です。コマンドラインで起動する際にノートブック単位で指定するので、JSMEを起動したノートブックとは別のもの(PanelChemistry_NGL.ipynb)を作っています。
アプリケーションにおける設定は上のJupyter notebook上で使う場合と同様です。viewerやsettings、flie_column、layoutといった変数の部分なのでそのまま引き継ぎます。
こんな感じで起動します。
# アプリケーションの見た目の色調設定 accent="#D2386C" # テンプレートを利用したレイアウトの作成 + servable() pn.template.FastListTemplate( site="Panel Chemistry", title="NGLViewer", sidebar=[file_column, settings, layout], main=[viewer], accent_base_color=accent, header_background=accent, ).servable();
あとはコンソールでコマンドを打ち込むだけです。
panel serve PanelChemistry_NGL.ipynb
ポートもデフォルト「5006」で変わらずなので「http://localhost:5006/PanelChemistry_NGL 」にブラウザでアクセスすればアプリを利用できます。

できました!起動時にファイルを指定しているので構造を読み込んでいますが、左上のファイルを選択から新しく構造を読み込むこともできます。
2-3. Py3DMolの場合
2-2-1. Jupyter notebook上で使う
最後にpy3Dmolの場合です。py3Dmolについては以下をご参照ください。
- Nicholas Rego and David Koes, 3Dmol.js: molecular visualization with WebGL, Bioinformatics (2015) 31 (8): 1322-1324 doi:10.1093/bioinformatics/btu829.
また、化学の新しいカタチさんの記事「py3Dmolを使って化学構造をJupyter上で美しく表示する」で詳細に解説してくださっています。
今までと同様にまずはライブラリを読み込みます。
import py3Dmol import panel as pn from panel_chemistry.pane import Py3DMol
pn.extension()
基本的な使い方は以下です。1行目py3DMol.view()のqueryを設定するとデータベースから構造をダウンロードして描画してくれます。下記ではmmtf(macromolecular transmission format)形式でPDB ID 1nktの構造をとってきています。
p = py3Dmol.view(query="mmtf:1nkt") p.setStyle({"cartoon": {"color": "spectrum"}}) pviewer=Py3DMol(p, height=400, sizing_mode="stretch_width", name="Basic") pviewer

できました!これ以外にもグリッド表示やインタラクティブなプロット(低分子の動くやつ)の例がGitHub上にありますが割愛します。
2-3-2. アプリケーションとして使う
つづいてアプリケーションとして使う場合です。ここでも新しいノートブックを(PanelChemistry_py3Dmol.ipynb)を作っています。
今までと同様、設定を書き込んだあと起動するだけです。
# 設定 background = pn.widgets.ColorPicker(value="#e6f6ff", name="Background") style=pn.widgets.RadioButtonGroup(value="sphere", options=["line", "cross", "stick", "sphere"], name="Style", button_type="success") # アプリケーションの見た目の色調設定 accent = "#0072B5" # テンプレートを利用したレイアウトの作成 + servable() pn.template.FastListTemplate( site="Panel Chemistry", title="Py3DMol Pane", sidebar=[background, style], main=[pviewer], header_background=accent, accent_base_color=accent ).servable();
あとはコンソールでコマンドを打ち込むだけです。
panel serve PanelChemistry_py3Dmol.ipynb
ポートもデフォルト「5006」で変わらずなので「http://localhost:5006/PanelChemistry_py3Dmol 」にブラウザでアクセスすればアプリを利用できます。

できました!
2-4. 3つのアプリを同時に
最後に、3つのアプリを同時に使う方法です。
それぞれのノートブックでservable()にした状態で、コマンドラインで以下の通り3つ並べて起動すればOKです。
panel serve PanelChemistry_JSME.ipynb PanelChemistry_NGL.ipynb PanelChemistry_py3Dmol.ipynb
この状態で「http://localhost:5006/ 」にアクセスすると以下のようになります。
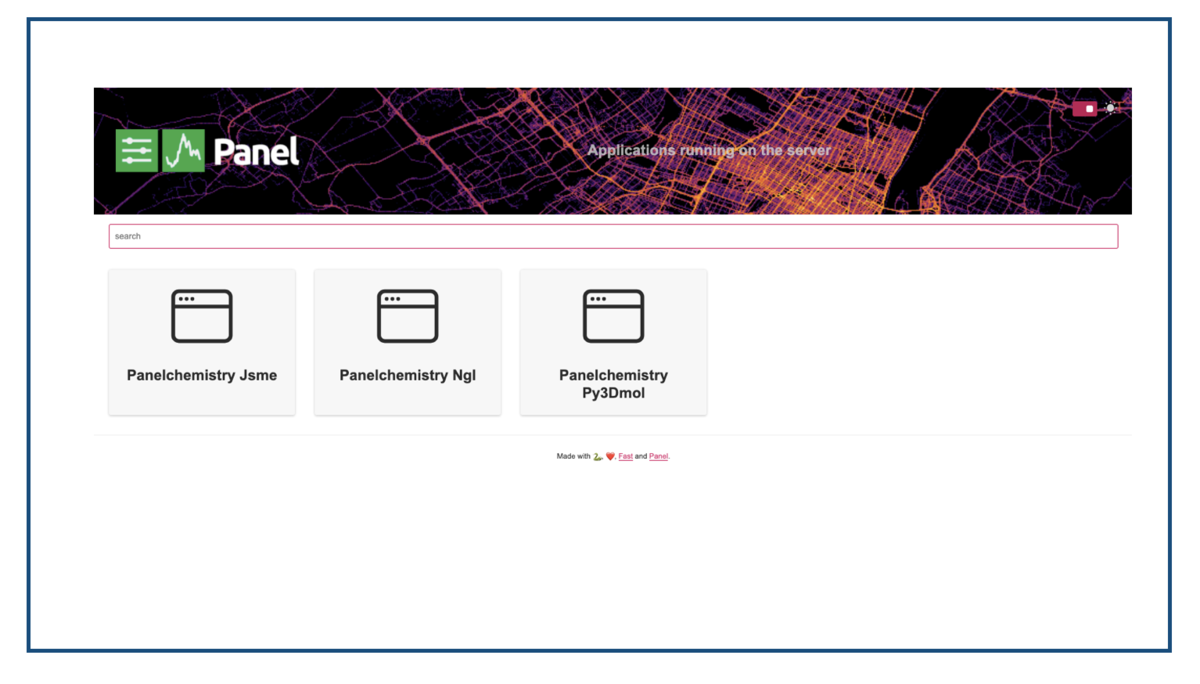
真ん中に並んでいるブロックがそれぞれJSME、NGL viewer、py3Dmolそれぞれのアプリに対応するので、クリックすれば開けます。もちろん別々のタブで同時に開くことも可能です。
3. おわりに
以上、今回は描画ツールPanel-Chemistryのご紹介でした。
化学構造式を描画・編集できるJSMEと、3Dの描画が得意なNGL Viewer、py3Dmolを並べて扱えるので便利ですね!一度コピー&ペーストしてノートブックを作っておけば、気軽にアプリとして起動できそうです。
まあ私はサーバーとかアプリとか全くわかってないんですけどね!
備忘録がわりのメモをはさんでいるのでGitHubのexampleよりわかりづらくなっているかもしれません。間違いがあったらご指摘いただけると嬉しいです。
ではでは!
Jupyter Notebookでも構造式を編集したい! 〜JSMEとipywidgetsでMolを扱う話〜
RDKitとJupyter notebookの組み合わせとても便利ですよね!「プログラミングなんてよくわからないぜ!」っていうへっぽこにとって結果が逐一見られるのは最高です!
ですが、遊んでいて一つ困ることが・・・「RDKitの構造式を書き変えるの面倒!!ちょっと変えたいだけなのに。。。」
というわけで今回はJupyter Notebook上で構造式を編集する方法を試してみたいと思います。具体的にはRDKitのMolオブジェクトからMolファイルで座標を取り出して、構造式を編集後、新しい座標から再度Molオブジェクトを作り直します。

- 1. JSMEとipywidgetを使おう
- 2. この記事で試す方針
- 3. RDKitで構造式を編集する正攻法
- 4. JSMEとくみあわせてRDKit Molを編集しよう
- 5. 試したけど上手くいかなかったこと
- 6. おわりに
1. JSMEとipywidgetを使おう
1-1. 先例 〜SMILESを取り出す方法〜
調べてみると同様のニーズはあるみたいで、優秀な方々が検討結果を共有してくださっていました。今回は、lithium0003さんのGitHubレポジトリ(JSME_ipywidget)を参考にさせていただくことにしました。*1
こちらの方法によると構造式エディタとしてJSMEを用い、Jupyter notebook上で実効するためipywidgetsを使うとのことでした。
1-2. JSMEって?
JSMEはフリーで利用可能な構造式エディタで、2013年に下記の論文で報告されています。*2
元々Javaで書かれていたJMEというエディタを、JavaScriptにアップデートしたもののようでBSDライセンスで配布されています。
Webページはこちら(→ JSME Molecule Editor)です。「Download the latest JSME distribution as zip file」をクリックすると最新版をダウロードできます。今回は少し古いバージョンを使います。

今回使うバージョンは2017-02-26です。最新版ではうまく動かなかったので参考例に近いバージョンにしました。

ダウンロードすると「JSME_2017-02-16ディレクトリ」が手に入りますが、このうち「jsmeディレクトリ」が今回必要となる部分です。これだけ作業ディレクトリに移しておきます。

なお、JSMEの見た目はこんな感じです。以前こちらのブログでも紹介したZINC20データベースでの利用例をもってきました。

ChemDrawやMarvinといった商用ソフトほどの機能はなさそうですが、十分な機能があり、何よりフリーで公開してくださっていることに感謝です。
1-3. 先例でコードのお勉強
lithium003さんの公開してくださっている方法はGitHubレポジトリのノートブック(smiles_draw.ipynb)の通りです。このノートブックでは「JSMEエディタをjupyter notebook上で使用し、描いた構造式のSMILESを取り出す」ということが行われています。
こんな感じ(READMEより)

でもさっぱりコードの中身がわからない!!Pythonも分からないのにJavaScriptも必要なの??
ということでGoogleで検索しながら順番に見ていきました。
全部で4つのセルからなります。
1-3-1. セル1
まず最初にipywidgetsの準備です。ipywidgetsはJupyter notebook上でインタラクティブなUIを手軽に作れるライブラリだそうです*3。ドキュメントはこちら(→ ipywidgetsドキュメンテーション)
とりあえずセル1のコードを引用します。ライブラリをインポートした後、SmilesEditorというクラスを作成しています。
from traitlets import Unicode, Bool, validate, TraitError from ipywidgets import DOMWidget, register @register class SmilesEditor(DOMWidget): _view_name = Unicode('SmilesView').tag(sync=True) _view_module = Unicode('smiles_widget').tag(sync=True) _view_module_version = Unicode('0.1.0').tag(sync=True) # Attributes value = Unicode('', help="SMILES value").tag(sync=True)
ここで行われているのはipywidgetsドキュメンテーションの「Low Level Widget TutorialのWidget skeleton項」 と「Building a Custom Widget - Email widgetのMaking the widget stateful項」を参考にすると様子がわかりそうです。
1行目のtraitletsはJupyter notebookで機能しているライブラリだそうで、Pythonの(動的に決まる)「クラスの属性の型をきちんと定めて、さらに細かいチェック機能を簡単に呼び出せる」ようにできるそうです*4。
2行目でipywidgetsライブラリからインポートしています。DOMWidgetのDOMはDocument Object Modelの略だと思います。DOMはJavaScriptの「表示されたWebサイトを動的に書き換えることができる」という特徴を支えている仕組みで、「HTMLをJavaScriptで操作することが出来る」そうです*5。
またregisterはビューが動的にアップデートするのに関わる関数のようです。*6
このセルの目的はJSMEを使えるようにするための箱(SmilesEditor)を準備することのようです。JavaScriptベースのJSMEを使うためのDOMWidgetのクラスSmilesEditorをつくっているんだと思います。たぶん。。。
1-3-2. セル2
セル2ではJSMEを使うための設定が書き込まれています。マジックコマンド%%javascriptでJupyter notebook上でJavaScriptが実行できるようにした上で処理が書かれています。
セル2を引用します。
%%javascript require.undef('smiles_widget'); require(['jsme/jsme.nocache.js']) define('smiles_widget', ["@jupyter-widgets/base"], function(widgets) { var SmilesView = widgets.DOMWidgetView.extend({ // Render the view. render: function() { this.smiles_input = document.createElement('div'); this.smiles_input.id = "jsme_container"; this.smiles_display = document.createElement('div'); this.smiles_display.textContent="SMILES : "; this.el.appendChild(this.smiles_display); this.el.appendChild(this.smiles_input); function myFunc(callback){ let jsmeApplet = new JSApplet.JSME("jsme_container", "480px", "480px"); jsmeApplet.setCallBack("AfterStructureModified", callback); }; setTimeout(myFunc, 500, this.smilesChanged.bind(this)); }, smilesChanged: function(jsmeEvent) { console.log(this, jsmeEvent) let jsme = jsmeEvent.src; let smiles = jsme.smiles(); this.smiles_display.textContent="SMILES : "+smiles; this.model.set('value', smiles); this.model.save_changes(); }, }); return { SmilesView: SmilesView }; });
冒頭2行のrequireは「一般的にモジュール化されたJavaScriptファイルを読み込む」ために用いられるものだそうです*7。
1行目のrequire.undef()はモジュールの定義を外すためのもので、内部の状態をリセットするための関数のようです*8。一度リセットしてから、2行目でJSME本体を呼びにいっている感じでしょうか??
2行目でJSMEを読み込むパスの通り、JSMEはノートブックと同じディレクトリに置かれた「jsmeディレクトリ」内の「jsme.nocache.jsファイル」を利用しています。requireはモジュール化されたJavaScriptファイルをよぶという説明通りですね。
つづいて3行目以降では具体的なSmilesViewの中身が書かれています。
thisというのは「JavaScriptに最初から用意されている特別な変数のこと」で、「呼び出した場所や方法によってその中身が変化する」という特徴があるそうです*9。よくわかりませんが、変数thisに色々格納して、JSME構造式描画ビューワー(smiles_input)やSMILES表示(smiles_display)の機能を設定している感じです。変数を設定した後appendChildメソッドを使って要素を実際に追加していく感じ*10。
準備ができたら、真ん中のfunctionから始まるブロックでJSEMのアプレットを読み込んでいます。AfterStructureModifiedというコールバックを使っているので、構造が編集されると変更後の構造を読み込んで反映する仕組みがつくられている感じです。・・・たぶん。
一番最後のブロックでは、構造式エディタに書かれた構造式からSMILESをとり出す仕組みが書かれているようです。letでは始まる2行はJSMEからSMILESの情報を取り出して、変数に格納している感じです*11。jsmeEvent.srcや.smiles()はJSME特有の箇所でJSME API ドキュメントなどに記載があります。
とりだしたSMILES情報をもとに、「this.smiles_display.textContent="SMILES : "+smiles;」ではインタラクティブな編集に応答してSMILESを表示する仕組みが、「this.model.set('value', smiles);」ではmodelのvalueというプロパティにSMILESを格納しています。
以上、セル2でJavaScriptのアプリ、JSMEを利用するための設定と書かれた構造からSMILESを取り出す仕組みの設定までが終わったようです。
1-3-3. セル3
セル3は、セル1とセル2で設定が完了したSmilesEditorを起動しています。
コードはシンプルです。
smiles = SmilesEditor() smiles

参考例のREADMEと同じものが立ち上がりました!
構造式を書き込むとEditor上部の「SMILES:」欄に対応するSMILES表記が反映されます。
1-3-4. セル4
セル4は構造式に対応するSMILESをJupyter notebookのセルに取り出すだけです。
コードはこちら
smiles.value
# 'OCc1ccccc1'
セル2でみたように、valueプロパティにSMILESを格納していたので.vlaueとすることでSMILESが取り出せました!
以上がlithium003さんが公開してくださっている方法です。
2. この記事で試す方針
コードを辿るだけで長くなってしまいましたが、今回私が行いたいのは「編集した構造式の新しい座標をベースにMolオブジェクトを作り直す」ことです。このためには「JSMEからSMILESではなくMolファイル形式の情報として編集結果を取得」すれば良さそうです。
具体的には以下を行います。
- RDKitのMolオブジェクトを
MolFileで出力する - JSMEをnotebook上で起動する
- MolFileを読み込んで構造式を編集
- 編集後の座標(Molファイル形式)を取得
- RDKit Molオブジェクトを作り直す(
MolFromMolBlock)
一時的にMolFileで出力するのが格好悪いですが、書き直すよりは楽かな???ということで。
3. RDKitで構造式を編集する正攻法
お試しの前に、RDKitで構造式を編集する真っ当な方法をご紹介しておきます。以下の日本語解説記事がとても参考になります。
①RWMolオブジェクトに変更して、②編集した後、③再度Molオブジェクトに戻すそうです。
例えばトルエンをベンジルアルコールにしようとするとこういう感じです。
from rdkit import Chem from rdkit.Chem import Draw # トルエンのMolオブジェクト toluene = Chem.MolFromSmiles("Cc1ccccc1") # 編集前後でindexを保つためにAtom Mapping for i, atom in enumerate(toluene.GetAtoms(), start=1): atom.SetAtomMapNum(i) Draw.MolToImage(toluene)

# RWMolオブジェクトに変換 rw_toluene = Chem.RWMol(toluene) # 酸素原子(Chem.Atom(8))を炭素原子(C:1)に追加して結合をつくる(AddBond) from_idx = rw_toluene.AddAtom(Chem.Atom(8)) to_idx = [atom.GetIdx() for atom in rw_toluene.GetAtoms() if atom.GetAtomMapNum() == 1][0] rw_toluene.GetAtomWithIdx(from_idx).SetAtomMapNum(8) rw_toluene.AddBond(from_idx, to_idx, Chem.BondType.SINGLE) # 編集後にMolオブジェクトに戻す benzylalcohol = rw_toluene.GetMol() Chem.SanitizeMol(benzylalcohol) Draw.MolToImage(benzylalcohol)
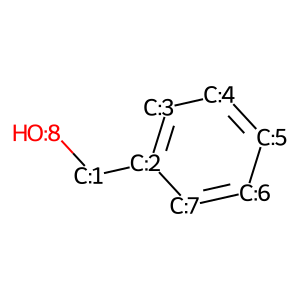
できました!けど面倒。。。分子ごとにatom indexを確認しないといけないのも一手間です。
4. JSMEとくみあわせてRDKit Molを編集しよう
では、JSMEと組み合わせて楽になるか???試してみましょう。
4-1. RDKitのMolオブジェクトから座標抽出
まずは、RDKitのMolオブジェクトから構造式の座標をMolファイル形式(temp.mol)で出力しておきます。
from rdkit import Chem # トルエンを作ってMolFileで出力する toluene = Chem.MolFromSmiles("Cc1ccccc1") Chem.MolToMolFile(toluene, 'temp.mol')
4-2. JSMEの設定(MolFile抽出)
JSMEを利用する箇所はほとんどlithium003さんのコードを利用させていただきます。ただし、ここで欲しいのは編集後のMol形式の座標なので、SMILESではなくMolFileを取り出す様に変更します。
目的にあわせて名前もSmilesEditorからMolEditorにしてみました。
準備のセル1はクラス名称等をかえていますが基本そのままです。
from traitlets import Unicode, Bool, validate, TraitError from ipywidgets import DOMWidget, register @register class MolEditor(DOMWidget): _view_name = Unicode('MolView').tag(sync=True) _view_module = Unicode('mol_widget').tag(sync=True) _view_module_version = Unicode('0.1.0').tag(sync=True) # Attributes value = Unicode('', help="Mol value").tag(sync=True)
JavaScriptのセル2では、よりシンプルにするためSMILES確認用ビューワーを削除しています。最後のブロックでMolFileを取り出すように変更しています。
%%javascript require.undef('mol_widget'); require(['jsme/jsme.nocache.js']) define('mol_widget', ["@jupyter-widgets/base"], function(widgets) { var MolView = widgets.DOMWidgetView.extend({ // Render the view. render: function() { this.mol_input = document.createElement('div'); this.mol_input.id = "jsme_container"; this.el.appendChild(this.mol_input); function myFunc(callback){ let jsmeApplet = new JSApplet.JSME("jsme_container", "480px", "480px"); jsmeApplet.setCallBack("AfterStructureModified", callback); }; setTimeout(myFunc, 500, this.molChanged.bind(this)); }, molChanged: function(jsmeEvent) { console.log(this, jsmeEvent) let jsme = jsmeEvent.src; let mol_data = jsme.molFile(); this.model.set('value', mol_data); this.model.save_changes(); }, }); return { MolView: MolView }; });
準備完了!
4-3. JSMEの実行と構造編集
JSMEは立ち上がるでしょうか?
MolEditorクラスをインスタンス化して実行します。
mol_editor = MolEditor() mol_editor

立ち上がりました!
右上の上下三角アイコンをクリックすると、構造式を種々の形式で読み書きできるプルダウンが開きます。
ここから「Paste Mol or SDF or SMILES」を選択すると新しい画面が開きます。「ファイルを選択」という箇所を選択するとファイルの読み込みができるので、先に出力しておいたMolファイル(temp.mol)を選択して取り込みます。「Accept」で描画スペースに構造式が反映されるので、あとは好きに編集しましょう!

適当に書き加えてみました。編集後の構造式の座標はvalueプロパティから無事取り出せるでしょうか???
print(mol_editor.value) """ Cc1ccc(C(C)O)nc1 JME 2017-02-26 Sat Jan 01 12:59:19 GMT+900 2022 10 10 0 0 0 0 0 0 0 0999 V2000 5.6001 1.2124 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 4.2000 1.2124 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 3.5000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 2.1000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 1.4000 1.2124 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 2.1000 2.4249 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 3.5000 2.4249 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0 6.3001 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 6.3001 2.4249 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0 0.0000 1.2124 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 1 2 1 0 0 0 0 2 3 2 0 0 0 0 3 4 1 0 0 0 0 4 5 2 0 0 0 0 5 6 1 0 0 0 0 6 7 2 0 0 0 0 7 2 1 0 0 0 0 1 8 1 0 0 0 0 1 9 1 0 0 0 0 5 10 1 0 0 0 0 M END """
できました!
最後の仕上げにこの情報からRDKit Molオブジェクトを再度作り直します。
# 編集後の構造式からMolオブジェクトを作る
modified_mol = Chem.MolFromMolBlock(mol_editor.value)
Draw.MolToImage(modified_mol)

再構築できました!
JSMEを使った構造式編集は、Atom indexとか考えなくても直感的に操作できるのでやりやすいですね!
5. 試したけど上手くいかなかったこと
やりたかったことは以上ですが、他にも試して上手くいかなかったことがいくつかあります。改善方法をご存知の方がいらっしゃったらご教示いただければ幸いです。
5-1. 失敗例1:JSMEバージョン依存
JSMEは継続的に開発されており、最新版は2021-07-13です。こちらを使って上記コードを試してみましたが上手くいきませんでした。
具体的にはJupyter notebok上にJSMEエディタ画面が立ち上がりませんでした。なので、今回は古いバージョンの2017-02-26を利用しています。
5-2. 失敗例2:Molファイルのクリップボードからのペースト
記事ではRDKitのMolオブジェクトを一度Molファイルとして出力し、JSMEにファイル読み込みという操作をしています。
「余計なファイルを増やすのは嫌だ」ということで、ファイル出力せずにMol情報だけコピー&ペーストしようとしました。
具体的にはpyperclipというライブラリを試しました。コード内でクリップボードにコピーしたりペーストしたりできるそうです*12。
pipでもcondaでもインストールできるそうなのでmambaしました*13。
mamba install -c conda-forge pyperclip
RDKitのMolオブジェクトからクリップボードへのコピーまでは以下の通りできました。
import pyperclip m = Chem.MolFromSmiles('CC') mb = Chem.MolToMolBlock(m) pyperclip.copy(mb)
このあと「Cntrol + V」すると、以下の通りペーストできます。
RDKit 2D
2 1 0 0 0 0 0 0 0 0999 V2000
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2990 0.7500 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
M END
問題はJSMEでペーストできなかったことです。Jupyter notebook上で起動したJSMEの「Paste Mol or SDF or SMILES」で枠内で「Cntrol + V」しましたが、全く反映されず書き込めませんでした。JSMEのテストページでは問題なく貼り付けと構造式の描画ができたので、私のコード(or Jupyter notebook上での挙動)に問題があるようです。
5-3. 失敗例3:JSME起動時に構造反映
最後に、全く方法がわからなかったのが「JSMEを起動する際に、編集したい元の構造式が書かれた状態で起動する」という方法です。
「構造式の読み込み作業が面倒だなー」と思ったので、RDKitのMolオブジェクトからMolBlockで取り出した情報を、JSME起動時に書き込みたかったのですが、ipywidgetもJavaScriptもよくわからないので無理でした。
6. おわりに
以上、今回は「Jupyter notebook上でも構造式を編集したい!」ということで、先行例を参考にJSMEとの連携を試してみました。
途中でふれた通りRDKitの構造編集はノンプログラマーの私にはハードルが高いです。「直感的に構造式を編集したい!」という点でエディターを使えるととても便利な気がします。
商用ソフトはお高そうなので個人では手を出しづらかったのですが、フリーでつかえるJSMEと組み合わせられることがわかってよかったです。開発・公開してくださっている方々に感謝です!
結局、コードの中身をあまり理解しないままコピー&ペーストしてしまいました。今回も間違いがたくさんありそうです。ご指摘、また改善方法等ご教示いただけると嬉しいです。
ではでは!!今年もよろしくお願いいたします!!
*1:他にもrdEditor(GitHubレポジトリ)というのがあって過去のRDKit UGMデモ発表があった様です。作成者の方のブログはこちら→rdEditor: An open-source molecular editor based using Python, PySide2 and RDKit
*2:B. Bienfait and P. Ertl, JSME: a free molecule editor in JavaScript, J. Cheminformatics 5:24 (2013)
*3:参考: Python: ipywidgets で Jupyter に簡単な UI を作る
*4:参考:jupyterを支える技術:traitlets(の解読を試みようとした話)
*5:参考:JavaScriptでDOMを操作する方法【初心者向け】
*6:参考:ipywidgetsドキュメンテーション Dynamic updateの説明より
*7:参考:【JavaScript入門】初心者がrequireの使い方で迷った時に読むまとめ!
*9:参考:thisって何?使い方を覚えて、JavaScriptをもっと楽しく使おう!
*10:参考:JavaScriptのappendChildメソッドの使い方を現役エンジニアが解説【初心者向け】
*12:参考:【Python】pyperclipでコピー&ペースト
*13:conda-forge pyperclip, BSD 3-clauseライセンス
Img2Molでピクトグラムを構造式に!
今年も残りわずかですね。2021年も色々なことがありました。
素人がお馬鹿をさらしているだけのブログですが、コメントいただけたり、twitterでリプライをいただいたりと皆様に暖かく接していただけて本当に感謝しています。
というわけで、今年最後のお遊びです!Img2Molをつかったピクトグラムの構造式化!!!
Img2Mol
以前こちらのブログでImg2Molというソフトウェアをご紹介しました。化学構造式の画像からOCRでSMILESを取り出すというものです。
ナノプシャン
ところで有機化学の分野には、化学構造式でお絵描きしてしまおう!面白い構造式の化合物を作ろう!という素敵な研究があります。
有名なところでは分子の小人、ナノプシャンがありますね!
ピクトグラム
2021年いろいろなことがありましたが東京オリンピックが印象的でしたね。賛否両論あったものの、この大変な状況で素晴らしいパフォーマンスを発揮されたスポーツ選手の皆様に勇気をもらった方も多かったのではないかと思います。
さて、オリンピックのでは開会式のパフォーマンス、ピクトグラムを再現!というのが話題になりましたね。
ピクトグラムを化学構造式に?
お馬鹿な私はこう思いました。
「ピクトグラムをImg2Molに投げ込んでめば、ピクトグラムを化学構造式で表現する方法がわかるのでは???」
Img2Molでオリジナルなナノプシャンをつくってやろうではありませんか!!
というわけで雑に作ったピクトグラムを投げ込んでみました!
はしれ
まずは走る人です。こんなピクトグラム作ってみました
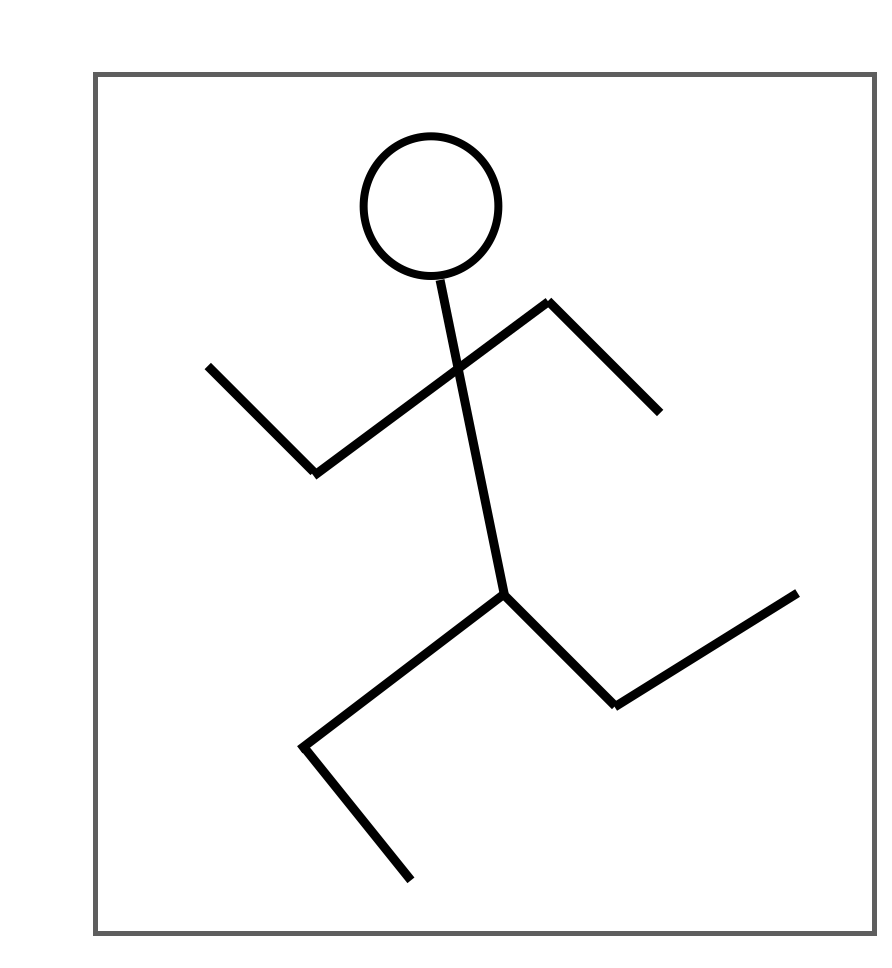
Img2Molによる解釈はこちら
足が表現されてる!宇宙人感!
ソーシャルディスタンス
距離を保つのが大事ですよね
構造式では?
Brはどこから??
レディオ体操
開催直前に色々ありましたが、小林賢太郎さんの所属するユニット ラーメンズのコント「バニー部」は独特の魅力がありますよね。
レディオ体操第一!
構造式では?
はじめて窒素原子がはいった!
おしまい
というわけでなんの中身もない記事でした。
こんな感じのくだらないブログですが、来年もこりずにお付き合いいただければ幸いです。
おしまい
TeachOpenCADD 2021をインストールした話
大幅にアップデートされたTeachOpenCADD 2021がリリースされました!
とりあえずインストールした!!
ちょっとつまずいた!!
のでインストール方法のメモです。
- 1. TeachOpenCADDって?
- 2. TeachOpenCADD 2021
- 3. アップデートはコンテンツだけじゃない!Webサイトも充実!
- 4. まだまだ増えてるコンテンツ
- 5. オンライン上で遊ぼう!(インストール不要)
- 6. ローカル環境で動かしたい!〜インストール①〜
- 7. もっとローカル環境で動かしたい! 〜インストール②〜
- 8. おしまい
1. TeachOpenCADDって?
TeachOpenCADDは「計算機支援ドラッグデザイン(CADD, computer-aided drug design)の基本的なタスクをどのように実行すれば良いか?」学習・教育するためのプラットフォームで、ケモインフォマティクスや構造バイオインフォマティクスについて学ぶ事ができる教材です。
Volkamer研究室が主体となって開発されており、オープンソース(CC BY 4.0)で公開されています。
2019年に公開・論文発表されており、以前こちらのブログでも取り上げさせていただきました。
今回、大幅にアップデートされたTeachOpenCADD 2021が公開されましたので改めてご紹介します。
2. TeachOpenCADD 2021
TeachOpenCADDはCADDのトピックをそれぞれ取り上げたトークトリアル(talk + tutorial = talktorial)と呼ばれる教材で構成されています。
2019年版ではT1 ~ T10の10回のトークトリアルでしたが、2021年版はT22までと大幅に内容がアップデートされています。
ChemRxivに論文(preprint)が公開されています。
- Sydow D, Rodríguez-Guerra J, Kimber TB, Schaller D, Taylor CJ, Chen Y, et al. TeachOpenCADD 2021: Open Source and FAIR Python Pipelines to Assist in Structural Bioinformatics and Cheminformatics Research. ChemRxiv
Figure 1. をご覧いただけば扱われている内容がつかめると思います。

すごい!!
もう少し概要を知りたい方はVolkamer Labのブログ(TeachOpenCADD 2021 release is out!)や、RDKit UGM2021でのご発表などもご参照いただくと良いかも。。。
- UGM2021のライトニングトーク(関連発表は9分50秒くらいからスタート(10分弱))
- プレゼンテーション資料へのリンク Dominique Sydow - TeachOpenCADD update
3. アップデートはコンテンツだけじゃない!Webサイトも充実!
盛りだくさんにコンテンツがアップデートされたTeachOpenCADDですが、2021のアップデートはさらに豪華です!
格好いいWebサイトまでできました!!

素敵だと思ったのは「Talktorials by collection」。関連するトークトリアルをグループ分けしたものが確認できます。

構造バイオインフォマティクスってインスタ映えしそうですね。
4. まだまだ増えてるコンテンツ
TeachOpenCADD 2021はトークトリアル T22までのアップデートと書きましたが、実はコンテンツは継続的に開発されていてWebサイト上では最新コンテンツも確認できます。
Volkamer Labのブログ(TeachOpenCADD Kinase edition is out!, 2021.12.14)によると、キナーゼに焦点を当てたスペシャルエディションができたそう。
Webサイトでチェックだ!

新しいトークトリアル(T23 ~ T28)がアップデート済みです!ますます充実していますね!
5. オンライン上で遊ぼう!(インストール不要)
TeachOpenCADDで遊ぶにはオンライン上で実行する方法と、ローカル環境にインストールする方法の2通りがあります。
オンラインはBinderで実行する形式で、セットアップ不要で手間要らずです。TeachOpenCADDのGitHub上にある「lauch / binder」ボタンをクリックするだけ!
BinderのURLは「 https://mybinder.org/v2/gh/volkamerlab/TeachOpenCADD/master 」なので、毎回GitHubに行くのが面倒な方はブックマークしておくと便利かもしれません。
こんな感じでBinderが起動して環境を準備してくれます。

準備にちょっと時間がかかります(5~10分くらい?)が、ワンクリックで使えるお手軽さは最高です。
準備できるとJupyter Labが立ち上がります。こんなの。
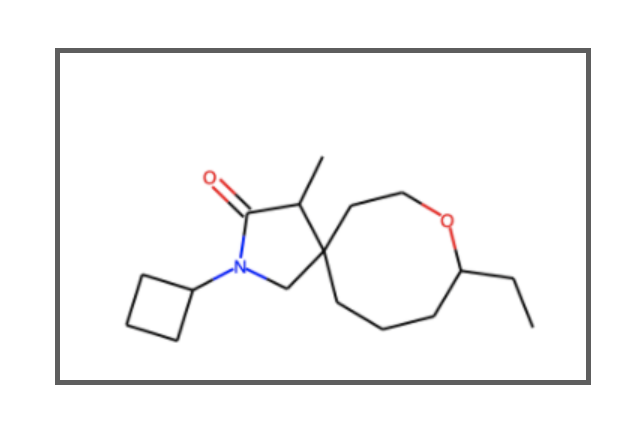
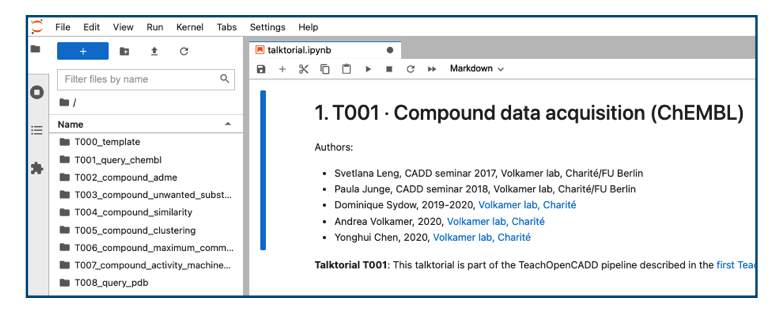
上図のように「teachopencaddディレクトリ → talktorialsディレクトリ → 見たいトークトリアルのディレクトリ(例:T001_query_chembl)」とたどって目的のノートブック(talktorkal.ipynb)を見つけます。
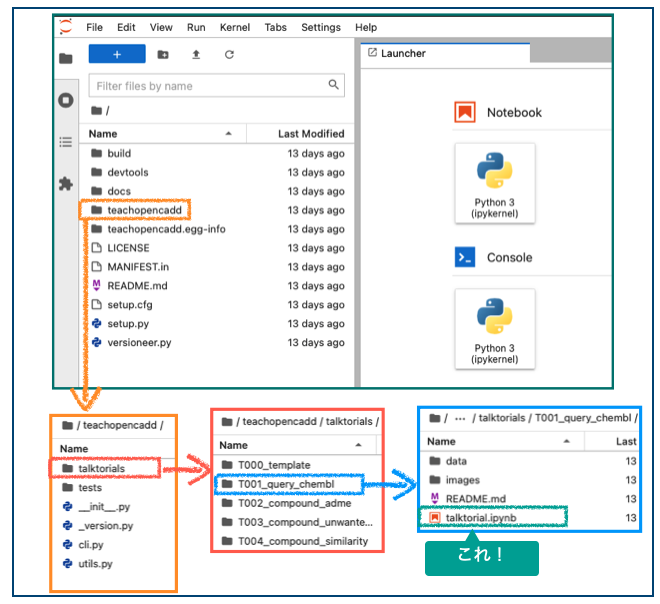
開くとWebサイト上で閲覧できるものと同じものが見られます。こんな感じ。
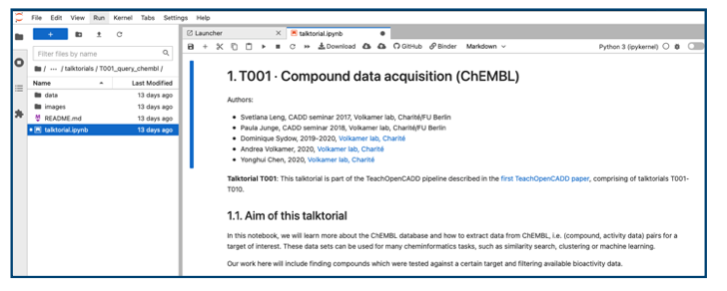
Webサイトとの違いは実際にノートブックのセルを実行できる事です。また編集できるので気になるコードを修正して結果の違いを見比べたり、試したい解析を追加したりと、いろんな遊び方ができそうです。
6. ローカル環境で動かしたい!〜インストール①〜
毎回Binderを待つのが嫌!オフラインでも試したい!という方はconda環境があればローカルにインストールすることもできます。
WebサイトのRun locallyページにインストール方法が書かれています。
Condaパッケージからのインストールは下記で、mambaを使った方法が書かれています。
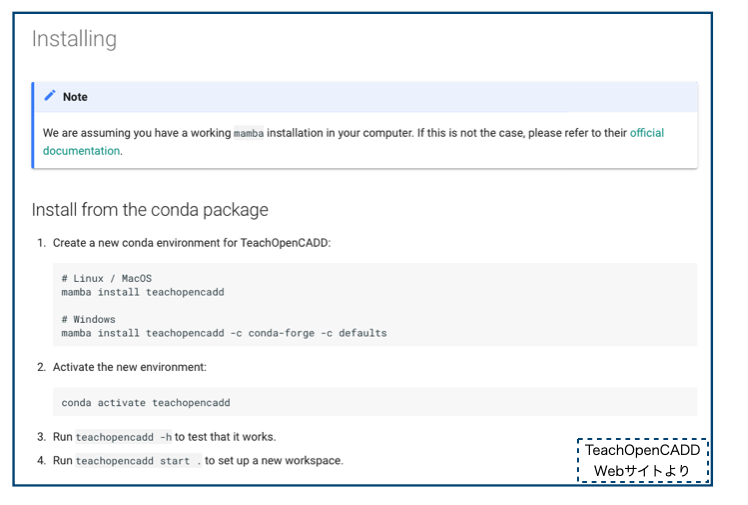
mambaは「C++で実装された高速なcondaパッケージマネージャー」だそうです*1。conda環境を使われているなら、mambaを追加インストールしておけば諸々のパッケージインストールが速くなるみたいです。(mambaオフィシャルドキュメント)
mambaは以下でインストールできます。私はMacなのでターミナルで以下を実行しました。
conda install mamba -c conda-forge
インストールできたら今まで「conda install ~~~」としてきたのを「mamba install ~~~」とするだけでOKです。
なお、TeachOpenCADDのインストール方法では新しい仮想環境をつくる工程が省略されているようです。通常通りconda create -n <仮想環境名>とすればOKですが、仮想環境の作成はmambaでもできるそうです。
やってみた!
# 仮想環境の作成 mamba create -n teachopencadd
できた!

上図の通りできた仮想環境(teachopencadd)のアクティベート、ディアクティベートの方法はcondaと変わらず同じコマンドです。
なお、mambaで作成した仮想環境もconda info -eで確認できる環境一覧に出てきますし、削除したくなったら以下でOKです。
conda remove -n teachopencadd --all
仮想環境ができたら、アクティベートしてTeachOpen CADDをインストールします。(Webサイトの順番だとできない気がするので逆にしてます)
# 仮想環境のアクティベート conda activate teachopencadd # TeachOpenCADDのインストール mamba install teachopencadd
インストールできたら以下のコマンドを実行しましょう!
teachopencadd start .
カレントディレクトリ(.)に新しいワークスペースが作成され、ターミナルにこんなのが表示されます。
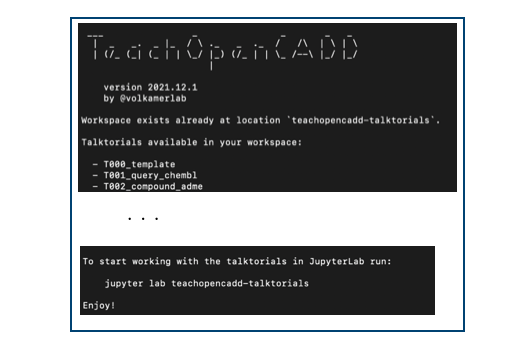
ロゴがかわいいですね。使えるトークトリアルの一覧とJupyterLabの起動コマンドが書かれています。
jupyter lab teachopencadd-talktorials
指示通り上記コマンドを実行すればBinderで確認したのと同じページがローカルで立ち上がります。
無事インストールできました!
なお、ワークスペースを作成する場所を指定したい時はteachopencadd start .コマンドのピリオドの部分にお好みのパスを入れてあげてください。
7. もっとローカル環境で動かしたい! 〜インストール②〜
「リリースを待ちきれない!」という方や、「自分で新しいトークトリアルを作るぜ!」という方は最新の開発版をインストールされるのも良いかもしれません。
一応ご紹介しておきます。
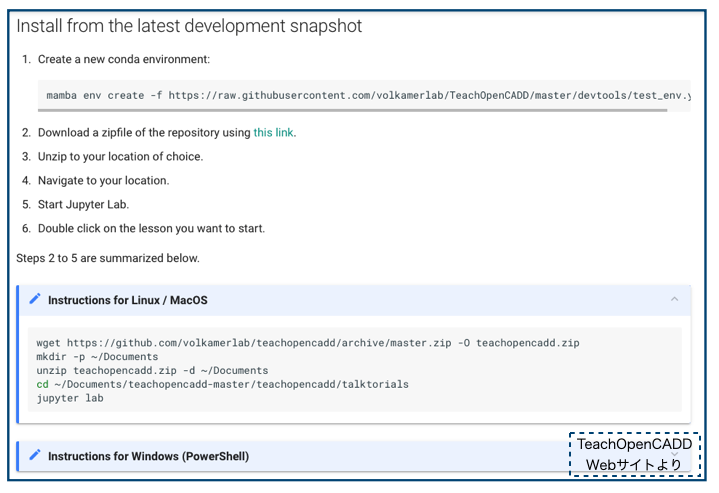
以下のコマンドでGitHubのTeachOpenCADDレポジトリに用意されているテスト環境用のtest_env.ymlを利用して仮想環境teachopencaddが作成されます。
mamba env create -f https://raw.githubusercontent.com/volkamerlab/TeachOpenCADD/master/devtools/test_env.yml
次に、以下でGitHubのレポジトリが作業ディレクトリ内にZIPファイルとしてダウンロードされます。
wget https://github.com/volkamerlab/teachopencadd/archive/master.zip -O teachopencadd.zip
unzipで解凍すればOKですが、以下では新しくDocumentsディレクトリを作ってその中に解凍しています。
# 作業ディレクトリ(test)の下にDocumentsディレクトリを作成 mkdir -p ./Documents # Documentsディレクトリ内にファイルを解凍 unzip teachopencadd.zip -d ./Documents
これでteachopencadd-masterディレクトリができました。この中のtalktorialsディレクトリに各トークトリアルのディレクトリが入っています。
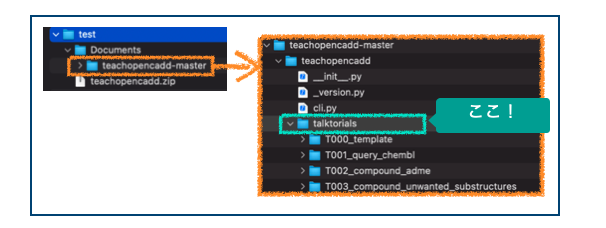
あとはtalktorialsディレクトリに移動してJupyterLabを立ち上げればおしまいです!仮想環境(teachopencadd)のアクティベートも忘れずに!
# talktorialsディレクトリに移動 cd ./Documents/teachopencadd-master/teachopencadd/talktorials # 仮想環境のアクティベート conda activate teachopencadd # JupyterLabの立ち上げ jupyter lab
mamba installしたときと同じJupyterLab画面が立ち上がりました!
先に見たインストール方法①ではワークスペース作成コマンドの実行があり、talktorialsフォルダができました。開発環境版を使う場合はワークスペース作成の代わりに、GitHubレポジトリを丸ごと持ってきてtalktorialsディレクトリをそのまま利用する、という違いがあるようです。
8. おしまい
以上、新しくパワーアップしたTeachOpenCADDについて簡単な紹介とインストール方法のご紹介でした。
コンテンツの大幅なアップデートだけでなく、Webサイトもとても見やすくなっていて素晴らしいですね!
Webサイトに書かれているインストール方法そのままだと、どうも私の環境(mac)ではうまくいかなかったので、ちょっとだけ手順をかえたものを記事にしてみました。色々と理解しないまま行っているので間違っているかもしれません。ご指摘いただけると嬉しいです。
なお、2021年版ではアップデートがなかったようですが、2019年版はKNIME版もありました。
- TeachOpenCADD-KNIME: A Teaching Platform for Computer-Aided Drug Design Using KNIME Workflows
J. Chem. Inf. Model. 2019, 59, 10, 4083–4086
こちらナイメストさんがnote上で使い方を解説してくださっています。
KNIMEの各ノードについて初歩から丁寧に説明してくださっていて、ほとんどKNIMEを使ったことのない私でも「ぽちぽちクリックしてたら動いたけど、背景ではこんなことが行われてるのねー」ととても勉強になるのでおすすめです。
正直、コードをみてもよくわからないのでKNIME版のアップデートが待ち遠しいです!!
なおなお、ついでのついでに皆様TalktorialsフォルダのトップがいずれもT000_templateとなっていることにお気づきになられましたか???
明日のTeachOpenCADDを作るのは君だ!!!
おしまい。